Поделиться


УЧЕБНОЕ ПОСОБИЕ
МДК 01.02. «ОТПУСК ЛЕКАРСТВЕННЫХ СРЕДСТВ И ТОВАРОВ
АПТЕЧНОГО АССОРТИМЕНТА»
РАЗДЕЛ 1. «ФАРМАЦЕВТИЧЕСКОЕ ТОВАРОВЕДЕНИЕ»
ДЛЯ СТУДЕНТОВ ПО СПЕЦИАЛЬНОСТИ ФАРМАЦИЯ
(Базовый и повышенный уровень среднего профессионального образования)
Грозный

Меркулова В.В.
УЧЕБНОЕ ПОСОБИЕ
МДК 01.02. «ОТПУСК ЛЕКАРСТВЕННЫХ СРЕДСТВ И ТОВАРОВ
АПТЕЧНОГО АССОРТИМЕНТА»
РАЗДЕЛ 1. «ФАРМАЦЕВТИЧЕСКОЕ ТОВАРОВЕДЕНИЕ»
ДЛЯ СТУДЕНТОВ ПО СПЕЦИАЛЬНОСТИ ФАРМАЦИЯ
(Базовый и повышенный уровень среднего профессионального образования)
Грозный
Учебное пособие МДК 01.02 «Отпуск лекарственных средств и товаров аптечного ассортимента» раздел 1 «Фармацевтическое товароведение» предназначен для студентов III курса очной и очно – заочной форм обучения по специальности Фармация (базовой и углубленной подготовки) и составлено в соответствии с рабочей программой МДК 01.02 «Отпуск лекарственных средств и товаров аптечного ассортимента».
|
Учебное пособие одобрено ЦМК профессиональных дисциплин Протокол № от Утверждено Методическим Советом колледжа Протокол № от |
|
Учебное пособие составлено в соответствии с ФГОС по специальности Фармация, базовая подготовка |
|
Автор: |
Меркулова Вера Владимировна, преподаватель профессиональных дисциплин высшей квалификационной категории Ивановского фармацевтического колледжа |
|
Рецензент: |
\
|
Учебное пособие МДК 01.02 «Отпуск лекарственных средств и товаров аптечного ассортимента» раздел 1 «Фармацевтическое товароведение», утверждено и рекомендовано Методическим советом Ивановского фармацевтического колледжа для использования в образовательном процессе по специальности Фармация
ОГЛАВЛЕНИЕ
|
Введение…………………………………………………………………………………….. |
6 |
|
Глава 1. Основы товароведения…………………….………….……………………….. |
7 |
|
Потребительские свойства товаров……………………………………………………. |
7 |
|
Глава 2. Товары аптечного ассортимента. Классификация и кодирование……… |
10 |
|
2.1. Классификация лекарственных средств и лекарственных препаратов…………….. |
10 |
|
2.2. Ассортимент медицинских фармацевтических товаров…………………………….. |
12 |
|
2.2.1. Парафармацевтические товары……………………………………………………... |
13 |
|
2.2.2. Общие требования, предъявляемые к качеству санитарно-гигиенических изделий и предметов ухода за больными…………………………………………………. |
16 |
|
2.3. Идентификация и штриховое кодирование товаров аптечного ассортимента……. |
16 |
|
2.4. Требования к нанесению кодов на потребительскую упаковку…………………….. |
18 |
|
2.5. Расчет контрольной цифры для кода EAN-13………………………………………... |
18 |
|
Глава 3. Требования к упаковке и маркировке лекарственных средств..………… |
19 |
|
3.1. Классификация упаковок……………………………………………………………… |
19 |
|
3.1.1. Классификация упаковки по назначению………………………………………….. |
19 |
|
3.1.2. Классификация упаковки по составу……………………………………………….. |
19 |
|
3.1.3. Классификация упаковки по применению…………………………………………. |
20 |
|
3.2. Требования к упаковке лекарственных средств……………………………………... |
21 |
|
3.3. Понятие маркировки. Производственные маркировочные надписи……………….. |
23 |
|
3.4. маркировка лекарственных средств…………………………………………………... |
25 |
|
3.5. Понятие и классификация товарных знаков…………………………………………. |
26 |
|
3.6. Знаки соответствия или качества……………………………………………………... |
27 |
|
Глава 4. Организация хранения лекарственных средств и других товаров аптечного ассортимента…………………………………………………………………. |
29 |
|
4.1. Общие требования к устройству и эксплуатации помещений хранения лекарственных средств…………………………………………………………………… |
29 |
|
4.2. Общие требования к помещениям для хранения лекарственных средств и организации их хранения……………………………………………………………… |
29 |
|
4.3. Особенности хранения отдельных групп лекарственных средств в зависимости от физических и физико – химических свойств, воздействия на них различных факторов внешней среды………………………………………………………………… |
30 |
|
4.3.1. Хранение лекарственных средств, требующих защиты от воздействия света…... |
30 |
|
4.3.2. Хранение лекарственных средств, требующих защиты от воздействия влаги….. |
31 |
|
4.3.3. Хранение лекарственных средств, требующих защиты от улетучивания и высыхания…………………………………………………………………………………… |
31 |
|
4.3.4. Хранение лекарственных средств, требующих защиты от воздействия повышенных температур………………………………………………………………… |
31 |
|
4.3.5. Хранение лекарственных средств, требующих защиты от воздействия пониженных температур………………………………………………………………… |
32 |
|
4.3.6. Хранение лекарственных средств, требующих защиты от воздействия газов, содержащихся в окружающей среде………………………………………………………. |
32 |
|
4.3.7. Хранение пахучих и красящих лекарственных средств…………………………… |
32 |
|
4.3.8. хранение дезинфицирующих лекарственных средств…………………………….. |
32 |
|
4.3.9. Хранение лекарственного растительного сырья…………………………………… |
32 |
|
4.3.10. Хранение медицинских пиявок……………………………………………………. |
33 |
|
4.4. Требования к помещениям для хранения огнеопасных и взрывоопасных лекарственных средств и организация их хранения……………………………………. |
33 |
|
4.4.1. Хранение огнеопасных лекарственных средств…………………………………… |
34 |
|
4.4.2. Хранение взрывоопасных лекарственных средств………………………………… |
35 |
|
4.5. Правила хранения медицинских изделий…………………………………………….. |
35 |
|
Глава 5. Система государственного контроля и сертификации лекарственных средств...…………………………………………………………………………………….. |
38 |
|
5.1. Государственный контроль качества лекарственных средств……………………… |
38 |
|
5.2. Виды государственного контроля…………………………………………………….. |
40 |
|
5.3. Порядок проведения сертификации качества лекарственных средств………….…. |
41 |
|
5.4. Процедура декларирования соответствия лекарственных средств…………………. |
45 |
|
5.5.Особенности декларирования лекарственных средств………………………………. |
46 |
|
Приложение ……………...…………………………………………………………….. |
48 |
ВВЕДЕНИЕ
В современных условиях развития фармацевтического бизнеса, большого числа производителей и поставщиков лекарственных средств (ЛС), значительного ассортимента ЛС, выявления на фармацевтическом рынке недоброкачественных и фальсифицированных ЛС сохраняет актуальность проблема качественной лекарственной помощи.
Политика всеобщего управления аптечным ассортиментом и его качеством, которая предполагает подход к организации с ориентацией на качество для достижения долгосрочного успеха путем удовлетворения потребностей потребителей и общества с учетом выгоды для коллектива, провозглашает всеобъемлющий контроль ЛС на всех этапах «жизненного цикла» с учетом одинаковой важности каждого этапа.
В настоящий момент вопросы оптимизации ассортимента, его качества и безопасности находятся под особым контролем государства, так как гарантия эффективности, безопасности и качества ЛС является важнейшей социальной задачей государства.
Цель данного учебного пособия – более полное и подробное ознакомление студентов с таким сложным и объемным разделом учебной программы как «Фармацевтическое товароведение».
Задачи:
1. Ознакомить студентов с разделом «Фармацевтическое товароведение»
2. Облегчить студентам процесс поиска теоретического материала по данному разделу программы;
3. Предоставить студентам возможность ознакомиться с нормативно – правовой базой по данной теме;
4. Подготовить студентов по теоретической части этой темы для написания дипломных работ по МДК 01.02 «Отпуск лекарственных средств и товаров аптечного ассортимента».
Учебное пособие состоит из 5 глав. Все главы посвящены теоретическому вопросу по обозначенной теме.
ГЛАВА 1. ОСНОВЫ ТОВАРОВЕДЕНИЯ
Товароведение –
наука об основополагающих характеристиках товаров, определяющих их
потребительские стоимости и факторах обеспечения этих характеристик.
Цель товароведения — изучение потребительских свойств
товаров, а также всех тех изменений, которые происходят в товаре на всех этапах
товародвижения. Товароведение как наука и учебная
дисциплина решает следующие основные задачи:
— систематизация множества товаров путем применения классификации, кодирования;
— разработка характеристик конкретных товаров, обусловливающих их потребительную ценность и способность удовлетворять определенные потребности человека, идентификация товаров, выявление фальсифицированных товаров;
— изучение ассортимента товаров и факторов, влияющих на его формирование;
— оценка качества товаров, выявление дефектов и причин их возникновения;
— товароведная характеристика конкретных товаров.
Товароведение - это наука о товаре. Естественно, что необходимо выяснить, что же такое товар.
1.1. Потребительские свойства товаров
Свойства товаров - это его объективная особенность, т.е. то, что отличает один товар от другого. Каждому товару присущи многие свойства, которые могут проявляться при его формировании, эксплуатации или потреблении.
Свойства товара, обусловливающие его полезность в процессе эксплуатации и потребления, называют потребительскими. Номенклатура потребительских свойств и их показателей определяется особенностями и назначением товара.
Потребительские свойства характерны для готовой продукции и товаров, реализуемых в розничной торговле.
Эргономические свойства товара - способность товара создавать ощущение удобства, комфортности, наиболее полного удовлетворения потребностей в соответствии с антропометрическими, физиологическими, психологическими и органолептическими характеристиками потребителя.
Антропометрические свойства товара - способность товаров при эксплуатации соответствовать измеряемым характеристикам потребителя. Эти свойства должны создавать комфортность, удобство при потреблении товаров.
Физиологические свойства товара - способность товаров обеспечивать удобство функционирования отдельных органов или частей тела человека при их использовании.
Психологические свойства товара - способность товаров обеспечивать при эксплуатации душевную комфортность. Психологические требования могут выражаться через восприятие вкуса, цвета, громкости и тембра звучания, яркости изображения. Восприятие отдельных пищевых продуктов в определенных регионах земного шара определяется национальными, религиозными, и др. признаками. (Например, мясо лягушек - мы не едим, мусульмане - не едят свинину).
При определении функциональных свойств товара нужно установить основное назначение товара, и условия использования по назначению.
1) свойства социального назначения - способность товаров удовлетворять индивидуальные и общественно- социальные потребности. Показатели: внешний вид, состав и содержание отдельных компонентов. (Например, количество ароматических веществ);
2) классификационное назначение - способность ряда свойств и показателей выступать в качестве классификационных признаков. Это: химический состав, отдельные вещества. (Например, содержание жира является классификационным признаком для жиросодержащих пищевых продуктов, т.к. творог бывает нежирный и жирный);
3) универсальное назначение- способность ряда свойств и показателей удовлетворять разнообразные потребности.
В зависимости от особенностей и удовлетворяемых потребностей потребительские свойства и показатели качества подразделяют на следующие группы:
1. назначения — функционального, социального, классификационного, универсального;
2. надежности — долговечность, безотказность, ремонтопригодность, сохраняемость;
3. эргономические — гигиенические, антропометрические, психологические, психофизиологические, эстетические;
4. безопасности — химическая, механическая, биологическая, радиационная, электрическая, магнитная, термическая, противопожарная.
Назначение — одно из определяющих потребительских свойств, характеризует способность товаров удовлетворять физиологические и социальные потребности, а также потребности в классификации товаров.
Надежность — свойство изделия сохранять во времени в установленных пределах все значения параметров, характеризующих способность изделия выполнять требуемые функции в заданных режимах и условиях.
Показателями надежности являются показатели безотказности, долговечности, ремонтопригодности и сохраняемости. По существу, показатели надежности дополняют характеристику товаров показателями функционального назначения, так как характеризуют продолжительность или полноту проявления эффекта от использования потребителем.
Эргономические свойства товаров характеризуют их приспособленность к использованию человеком в производственных и бытовых процессах. К эргономическим свойствам и показателям относятся гигиенические, антропометрические, психофизиологические и психологические.
Гигиенические — свойства товаров, влияющие на организм и работоспособность человека. Гигиенические свойства определяются условиями эксплуатации изделия: температурой и влажностью воздуха, шумом, вибрацией и другими, а также природой материала.
Антропометрические свойства — способность изделия или его деталей соответствовать размерам, форме и массе потребителя. Показатели антропометрических свойств: размеры одежды, обуви, мебели; форма посуды; размеры и форма бытовой техники и т. п.
Психофизиологические свойства — способность товаров обеспечивать соответствие особенностям органов чувств человека: зрительных, слуховых, обонятельных, осязательных, вкусовых. Психологические свойства — способность товаров соответствовать психике потребителя (восприятию, мышлению и памяти).
Эстетические свойства — способность выражать чувственно воспринимаемые признаки социально-культурной значимости товаров, степени их полезности и целесообразности, технического совершенства. К показателям эстетических свойств относят: форму изделия, цвет, ценностность композиции, стиль, моду, оригинальность изделия.
Экологические свойства характеризуют степень вредного воздействия продукции на окружающую среду, возникающего при производстве, потреблении или эксплуатации товаров, а также при их хранении и утилизации.
Свойства безопасности потребления — это обеспечение биологической, механической, электрической, пожарной и других видов безопасности при эксплуатации или потреблении товаров. В стандартах предусматриваются обязательные требования, обеспечивающие безопасность. На товары, использование которых по истечении определенного срока представляет опасность, должны устанавливаться сроки годности.
ГЛАВА 2. ТОВАРЫ АПТЕЧНОГО АССОРТИМЕНТА.
КЛАССИФИКАЦИЯ И КОДИРОВАНИЕ
Основные понятия, термины и определения, которые используются при классификации товаров в любой сфере, в том числе и в фармацевтическом товароведении, установлены ГОСТ 6.01.1-87 «Единая система классификации и кодирования технико-экономической информации. Основные положения».
В фармацевтическом товароведении объектом классификации являются фармацевтические товары, их свойства и показатели качества. Объектами также является сырье и материалы для производства фармацевтических товаров, методы оценки качества и виды контроля качества фармацевтических товаров.
В фармацевтическом товароведении объектом классификации являются фармацевтические товары, их свойства и показатели качества. Объектами также является сырье и материалы для производства фармацевтических товаров, методы оценки качества и виды контроля качества фармацевтических товаров.
Классификация товаров необходима для выполнения следующих целей:
- для автоматизированной обработки информации о лекарственных средствах и других товарах аптечного ассортимента;
- для изучения потребительских свойств и качества товара;
- для учета, а также планирования товарооборота аптечных организаций;
- для совершенствования системы стандартизации товаров фармацевтического рынка;
- для составления реестров цен и каталогов, а также прайс-листов;
- для проведения маркетинговых исследований на фармацевтическом рынке;
- при сертификации продукции;
- для проведения статистического анализа производства, реализации и использования продукции на различных уровнях, в том числе на макроэкономическом, региональном и отраслевом. Классификация фармацевтических товаров обладает высокой специфичностью и соответственно должна отвечать определенным требованиям.
2.1. Классификация лекарственных средств и лекарственных препаратов
|
Самостоятельные классификации лекарственных средств и лекарственных препаратов: 1. Классификации, ориентированные на медицину. 2. Многофункциональные классификации. 3. Классификации, ориентированные на фармацию. 4. Прочие классификации. Классификации в составе других классификаций: 1. Международная классификация изобретений (МКИ). 2. Международная классификация товаров и услуг (МКТУ). 3. Общероссийский классификатор продукции (ОКП). 4. Прочие классификации. 5. По покупательскому спросу:
- товары повседневного спроса; - товары предварительного выбора; - товары особого спроса; - товары пассивного спроса. По характеру спроса фармацевтические товары являются товарами массового спроса. Тем не менее, в группе фармацевтических товаров можно различить товары массового спроса и товары пассивного спроса. Чаще всего в группу товаров пассивного спроса попадают товары либо имеющие высокую цену, либо обладающие низкими качественными характеристиками, либо по этом товаре у потребителей очень мало информации, а также почти отсутствует реклама. К товарам особого спроса — относят товары, обладающие уникальными свойствами, для приобретения которых требуются дополнительные усилия и затраты со стороны потребителей. Выделяют товары выборочного спроса. Приобретение такого товара связано с предварительной оценкой имеющегося ассортимента и последующим выбором товара в результате сравнения по таким категориям, как качество, оформление, цена, фирма-производитель, и соответственно, страна – производитель. АТС классификация. Первые международные исследования в области потребления ЛС были проведены в 60-е годы. Однако каждое из них базировалось на собственной классификации. Для получения сопоставимой информации были необходимы научно обоснованные методологические стандарты. В 1969 г в Осло под эгидой ВОЗ прошел симпозиум по проблемам потребления ЛС, где было принято решение о необходимости разработки международной классификационной системы для изучения особенностей потребления ЛС. В 1974 г. Норвежское агентство по контролю за ЛС, опираясь на анатомо-терапевтическую классификацию, разработанную Европейской ассоциацией фармацевтических рыночных исследований, создало систему, известную в настоящее время как классификационная система АТС (Анатомо-Терапевтическая Система классификации). В 1996 г. ВОЗ рекомендовала использование системы АТС (точнее, ATC/DDD, последние три буквы означают «установленные суточные дозы» — Defined Daily Doses) в качестве международного стандарта при проведении исследований по потреблению лекарственных препаратов. Сегодня единственными странами, не принявшими данную классификационную систему, является США и Канада. В классификационной системе АТС лекарственные препараты разделяются на группы в зависимости от их действия на определенный анатомический орган или систему, а также от их химических, фармакологических и терапевтических свойств. На этой основе каждому препарату присваивается отдельный код. По рекомендациям ВОЗ его структура состоит из 5 уровней. 1 уровень — это 14 основных анатомических групп (классов), обозначаемых одной латинской буквой, которая в коде препарата стоит первой. Группы первого уровня разделены на группы 2 уровня (основные терапевтико-фармакологические), различающиеся, как правило, по основному терапевтическому применению. Они обозначаются двумя арабскими цифрами. 3 уровень классификации (терапевтико-фармакологические подгруппы) обозначается одной латинской буквой. 4 уровень представлен терапевтико-фармаколого-химическими группами, которые также обозначаются одной латинской буквой. 5-й уровень отражает химические субстанции и передается двумя цифрами. Кодировка препарата Фуросемид АТС: 1-й уровень, основная анатомическая группа: С – Сердечно-сосудистая система 2-й уровень, основная терапевтическая группа: 03 - Мочегонные средства 3-й уровень, терапевтическая группа: С - Высокоактивные диуретики 4-й уровень, химико-терапевтическая подгруппа: А - Простые препараты сульфаниламидов 5-й уровень, химические субстанции: 01 - Фуросемид Весь код: С03СА01 Главные цели ВОЗ - применение данной классификации в международных исследованиях потребления лекарств и для мониторинга побочных эффектов. Это позволяет использовать информационные системы по лекарственным препаратам для выбора альтернативных средств, а также для оценки лекарственных взаимодействий, предупреждения дублирования в назначении препаратов, контроля правильного выбора дозы лекарственного средства.
2.2. Ассортимент медицинских фармацевтических товаров |
Товары основного ассортимента, традиционно отпускаемые из аптечных организаций, составляют основу перечня жизненно необходимых лекарственных средств и препаратов и называются фармацевтическими.
Фармацевтическими товарами или товарами основного ассортимента являются:
1. лекарственные средства, в том числе и гомеопатические:
- лекарственные средства, отпускаемые по рецепту врача;
- лекарственные средства, отпускаемые без рецепта;
2. изделия медицинского назначения (ИМН):
- перевязочные средства;
- предметы ухода за больными;
- другие ИМН.
Кроме того, существует такое понятие, как тип лекарственного средства. Тип лекарственного средства — это группа медикаментов с устойчивым сочетанием определенных товарных свойств, главное па которых служит термином — наименованием данной группы.
Рассмотрим основные типы лекарственных средств.
1. Классические, гомеопатические. В качестве типологического признака — система медицины. При анализе рынка чаще всего принимается во внимание только тип «гомеопатические лекарственные средства».
2. Лечебные, профилактические и диагностические лекарственные средства. В качестве типологического признака — предназначение. Типология очевидная, но при анализе рынка ЛС практически не используется.
3. Готовые, экстемпоральные лекарственные средства, внутриаптечная заготовка. Типологический признак — характер производства.
4. Оригинальные, воспроизводимые лекарственные средства. Типологический признак — первичность производства. При анализе рынка необходимо определить типологический признак во избежание терминологической путаницы.
5. Патентованные, непатентованные лекарственные средства. Типологический признак — правовая защита. При анализе рынка необходимо определить типологический признак но избежание терминологической путаницы.
6. Новые, известные лекарственные средства. Типологический признак — новизна. При анализе рынка обычно принимается во внимание только тип «новые лекарственные средства».
7. Стандартные, бракованные лекарственные средства. Типологический признак - качество. При анализе рынка обычно принимается во внимание только тип «бракованные».
8. Подлинные, фальшивые лекарственные средства. Типологический признак - подлинность. При анализе рынка обычно принимается во внимание только тип «фальшивые».
9. Рецептурные, безрецептурные лекарственные средства. Типологический признак - вид отпуска.
2.2.1. Парафармацевтические товары
Товары
дополнительного ассортимента
Парафармацевтическая продукция — это товары дополнительного аптечного ассортимента,
сопутствующие лекарственные средства и изделия медицинского назначения,
предназначенные для профилактики, лечения заболеваний, облегчения состояния
человека, ухода за частями тела (Рис. 1). Впервые понятие парафармацевтической
продукции (ПФП) было введено в 1994г. Минздравмедпромом.
 |
 |
Рис. 1. Классификация товаров дополнительного ассортимента
Товары, предназначенные для матери и ребенка. В эту группу включены белье специализированное женское (для беременных, кормящих женщин); бандажи, пояса дородовые и послеродовые; изделия или приспособления для мытья ребенка; комплекты для сцеживания молока; памперсы, подгузники, пеленки; средства гигиены для новорожденных и их матерей и др. Товары для перевязки и фиксации. Сюда входят перевязочные средства различных видов, лечебные гольфы, чулки, колготки; лейкопластыри разных размеров и видов, повязки медицинские разных типов и назначений и др.
Бинты
Перевязочные
средства
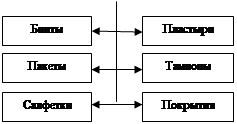 |
 |
||
Рис. 2. Классификация товаров ухода за больными
Предметы ухода за больными – это парафармацевтические товары, обеспечивающие надлежащее обслуживание больных (Рис. 2), облегчающие их страдания и предотвращающие осложнения в процессе лечения, а похоже создающие оптимальные условия в процессе проведения различного рода медицинских и других процедур. Это дозаторы лекарств, противопролежневые матрацы, простыни, калоприемники, мочеприемники и др.
Товары для контроля показателей здоровья. В эту группу входят различные приборы (средства, аппараты) медицинского назначения для измерения давления, пульса, силы, энергии, температуры тела; экспресс-тесты для диагностики ранней беременности, тесты на овуляцию, для выявления наркотиков, для диагностики инфекционных заболеваний (гепатит, сифилис, СПИД и др.), острого инфаркта миокарда, ряда онкологических заболеваний и др. средства контроля (тест-полоски) разных типов (определение холестерина, глюкозы в крови и др.) и др.
Товары для пропаганды здорового образа жизни. Сюда включают медицинскую, фармацевтическую, санитарно-просветительную, спортивную литературу, газеты, журналы, видео- и аудиопродукцию и предметы для обеспечения здорового образ жизни (тренажеры, массажеры и др.).
Санитарно - гигиенические изделия - это парафармацевтические товары обусловливающие соблюдение в домашних условиях и ЛПУ установленных санитарных правил, проведение различных мероприятий направленных на улучшение и оздоровление условий жизни и труда человека (Рис. 3).
 |
Рис. 3. Санитарно – гигиенические изделия
Классификация санитарно-гигиенических изделий и предметов ухода за больными:
1. По материалу для изготовления:
- изделия из резины (грелки медицинские, пузыри для льда, трубки медицинские, спринцовки типа А и др.) (Рис. 4);
![]()

![]()
Рис. 4. Виды спринцовок
- изделия из латекса (перчатки медицинские, напальчники, соски детские);
- изделия из пластмассы (наконечники клизменные, стаканчики для приема лекарств, судна подкладные, мочеприемники);
- изделия из стекла (наконечники влагалищные, мочеприемники, стаканчики для приема лекарств, банки медицинские и др.);
- изделия из металла (судна подкладные, поильники, тазики почкообразные. костыли и трости опорные);
- изделия из фарфора (судна подкладные, поильники);
- изделия из древесины (костыли и трости опорные);
- изделия из текстильных материалов (бандажи, корректоры осанки, шорты антицеллюлитные, носки противогрибковые);
- изделия из замши (бандажи грыжевые двусторонние и др.);
- комбинированные изделия (резина и стекло: молокоотсосы ручные; резина и пластмасса: спринцовки типа Б; резина и ткань: клеенки медицинские резинотканевые; стекло и латекс: пипетки глазные; пластмасса и текстиль: зубочистки с нитью).
2. По области применения:
- для терапевтических целей (грелки медицинские, пузыри для льда, банки медицинские, трубки медицинские, пояса послеоперационные и др.); для профилактических целей (бандажи лечебные, молокоотсосы, круги подкладные, корректоры осанки, колготки и чулки антиварикозные, шорты антицеллюлитные, подпяточники, маски и др.);
- для диагностических целей (катетеры, зонды, трубки медицинские и др.); для гигиенических целей (губки туалетные, щетки для рук, ватные шарики, ватные диски и др.);
- для дозирования жидких лекарственных средств (пипетки глазные, стаканчики для приема лекарств, поильники);
- для передвижения и создания опоры (костыли и трости опорные, костыли локтевые);
- для коррекции костно-суставной системы (супинаторы, корректоры осанки и др.);
- для ухода за лежачими больными (судна подкладные, мочеприемники, калоприемники, поильники, спринцовки, клеенка медицинская и др.); для ухода за новорожденными (соски молочные детские, соски-пустышки, палочки гигиенические, кольца зубные, аспираторы назальные и др.); для защиты рук медицинского персонала (перчатки медицинские, напальчники);
- для ухода за зубами (щетки зубные, зубочистки, зубочистки с нитью и стимулятором десен, нити зубные);
- для гигиены женщин (прокладки, тампоны и салфетки гигиенические);
- для проведения лабораторных работ (пипетки глазные, спринцовки, трубки медицинские, баллоны и мехи резиновые и др.);
- для комплектования медицинской аппаратуры (трубки медицинские, баллоны и мехи резиновые).
2.2.2. Общие требования, предъявляемые к качеству санитарно-гигиенических изделий и предметов ухода за больными
- соответствие материалу изготовления;
- соответствие размерам (объему, длине, ширине, диаметру) и геометрической форме;
- отсутствие внешних дефектов (вмятин, царапин, механических повреждений, разрывов, наплывов, разнотона и т. д.) на самом изделии и его упаковке;
- герметичность и прочность (для 1релок резиновых, пузырей для льда, кругов подкладных, кружек ирригаторных, перчаток хирургических);
- эластичность (для бандажей лечебных, трубок медицинских, сосок детских корректоров осанки и др.);
- упругость (для спринцовок, колец маточных);
- неслипаемость внутренней поверхности изделий (для изделий из резины I латекса);
- комплектность (для изделий, включающих два и более конструктивных элементов);
- наличие и полнота маркировки;
- стойкость к моющим и дезинфицирующим агентам.
2.3. Идентификация и штриховое кодирование товаров аптечного ассортимента
Данный раздел пособия представлен Методическими указаниями 64-802-00 «Система классификации и кодирования химфармпродукции. Общие правила штрихового кодирования лекарственных средств». Данный документ введен в действие Информационным письмом N 15-386 от 09.11.2000.
· ГОСТ Р 51201-98. "Автоматическая идентификация. Штриховое кодирование. Требования к символике "EAN/ЮпиСи"
· ГОСТ 16330-85. Система обработки информации. Шрифты для оптического чтения. Типы, основные параметры и размеры
· МУ 9467-015-05749470-98. "Графическое оформление лекарственных средств. Общие требования".
Потребительская тара - тара, поступающая к потребителю с лекарственными средствами и не выполняющая функцию транспортной тары.
Дополнительный символ - символ, применяемый для кодирования дополнительной информации, не содержащейся в основном символе.
Четный паритет - характеристика кодирования знака символа, указывающая, содержит ли он четное число темных модулей - штрихов (сумма модулей штрихов знака четного паритета является четным числом).
Знак-ограничитель - комбинация штрихов/пробелов, служит для разделения основного символа на две половины.
Организация, ответственная за присвоение номера - организация, присоединившаяся к ЕАН, отвечающая за управление системой ЕАН и зарезервированные номера в пределах своей территории.
Набор знаков - серия комбинаций штрихов и пробелов с четным или нечетным паритетом, кодирующая цифры от 0 до 9.
Нечетный паритет - характеристика кодирования знака символа, указывающая, содержит ли он нечетное число темных модулей - штрихов (сумма модулей штрихов знака нечетного паритета является нечетным числом).
Лекарственные средства в потребительской таре должны иметь идентификационный штриховой код ЕАН-13 или ЕАН-8, наносимые на потребительскую тару в соответствии с действующим стандартом Международной Ассоциации ЕАН.
Идентификационный штриховой код должен состоять из штрихового кода (символа) и уникального (цифрового) номера.
Для потребительской тары и этикеток, имеющих малые площади размещения информации, на которых трудно или невозможно разместить код ЕАН-13, должен применяться символ ЕАН-8.
Штриховой код должен иметь прямоугольную форму, образованную последовательностью светлых и темных параллельных штрихов, и светлое поле со всех сторон.
Штриховой код должен быть выполнен так, чтобы он мог считываться сканирующим устройством в обоих направлениях.
Формат и структура символов ЕАН-13:
Символ ЕАН-13 составлен следующим образом (при считывании слева направо):
· - типовой знак-ограничитель;
· - 6 информационных знаков символа из набора знаков нечетного и четного паритета;
· - центральный знак-ограничитель;
· - 6 информационных знаков символа из набора знаков четного паритета;
· - типовой знак-ограничитель.
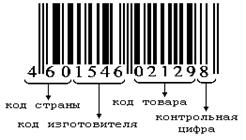 Последний информационный
знак символа кодирует контрольный знак. Так символ ЕАН-13 содержит только 12
информационных знаков символа, но кодирует 13 цифр данных (включая контрольный
знак). Сумма модулей (штрихов) знака определяет его паритет (четный или нечетный).
Пример символа штрихового кода ЕАН-13 приведен на рисунке 5.
Последний информационный
знак символа кодирует контрольный знак. Так символ ЕАН-13 содержит только 12
информационных знаков символа, но кодирует 13 цифр данных (включая контрольный
знак). Сумма модулей (штрихов) знака определяет его паритет (четный или нечетный).
Пример символа штрихового кода ЕАН-13 приведен на рисунке 5.
На потребительскую тару лекарственных средств штриховой код должен быть нанесен в соответствии с требованиями МУ 9467-015-05749470.
Место размещения кода ЕАН определяется конструкцией и размером потребительской упаковки, применяемой для упаковывания лекарственных средств.
Код ЕАН должен наноситься на достаточно гладкую и ровную поверхность. Не допускается наносить код на швы, перегибы, складки, морщины, места склейки и другие неровности на потребительской таре.
Код ЕАН может иметь как горизонтальное, так и вертикальное расположение по отношению к основанию (дну) тары, флакона, банки и т.п.
Код ЕАН рекомендуется наносить на белый фон, допускается желтый, оранжевый и светло-коричневый.
На каждую потребительскую упаковку должен быть нанесен только один код ЕАН.
1. Складываем цифры,
стоящие на четных позициях, затем на нечетных позициях:
|
Номер позиции |
1 |
2 |
3 |
4 |
5 |
6 |
7 |
8 |
9 |
10 |
11 |
12 |
13 |
|
Значение |
4 |
0 |
1 |
8 |
9 |
9 |
3 |
4 |
0 |
4 |
7 |
8 |
7 |
|
Четные |
|
0 |
+ |
8 |
+ |
9 |
+ |
4 |
+ |
4 |
+ |
8 |
=33 |
|
Нечетные |
4 |
+ |
1 |
+ |
9 |
+ |
3 |
+ |
0 |
+ |
7 |
|
=24 |
2. Теперь складываем
результат сложения цифр на четных позициях, помноженный на три и результат
сложения цифр на нечетных позициях:
(33*3) + 24
= 123
3. Затем от 123 отделяем последнюю цифру – 3 и вычитаем её из 10 (константа) всегда 10 – 3 = 7
4. Полученная в результате последняя семерка и соответствует контрольной цифре.
ГЛАВА 3. ТРЕБОВАНИЯ К УПАКОВКЕ И МАРКИРОВКЕ
ЛЕКАРСТВЕННЫХ СРЕДСТВ
3.1. Классификация упаковок
В товароведении существуют разные классификации упаковки. Для медицинских и фармацевтических товаров упаковку можно классифицировать по следующим признакам:
1) по назначению (Рис. 6);
2) по составу;
Упаковка
3)
по применению.
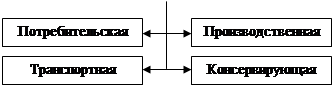 |
Рис. 6. Классификация упаковки по назначению
3.1.1. Классификация упаковки по назначению
По признаку назначения упаковка подразделяется на потребительскую, транспортную, производственную и консервирующую.
Потребительская упаковка попадает с продукцией непосредственно к потребителю, является неотъемлемой частью товара и входит в его стоимость. Такая упаковка не предназначена, как правило, для самостоятельного транспортирования, имеет ограниченную массу, вместимость и размеры.
Транспортная упаковка составляет отдельную самостоятельную транспортную единицу и используется для перевозки товаров в потребительской упаковке или неупакованной продукции.
Производственная упаковка используется как часть технологии при организации производственного процесса на одном или нескольких предприятиях и не предназначена для реализации продукции в розничной торговой сети.
Консервирующая упаковка необходима для долгосрочного сохранения сырья, материалов, изделий, техники, а также опасных отходов (химических, радиоактивных и т.д.).
3.1.2. Классификация упаковки по составу
По составу различают два вида упаковки: тара и вспомогательные упаковочные средства (Рис. 7).

Рис. 7. Классификация упаковки по составу
Тара является наиболее важным, а иногда и единственным элементом упаковки, который представляет собой изделие для размещения продукции, выполненное в виде замкнутого или открытого корпуса. Тара осуществляет функции упаковки самостоятельно или в сочетании со вспомогательными упаковочными средствами, которые являются другими элементами упаковки.
К вспомогательным упаковочным средствам, которые используются в потребительской и транспортной упаковке, относятся: укупорочные средства, этикетки, покрытия, обертки, герметизирующие, скрепляющие и амортизирующие элементы, вещества, которые создают защитную атмосферу внутри упаковки.
3.1.3. Классификация упаковки по применению
Упаковка
По
признаку применения упаковка подразделяется на первичную, вторичную и третичную
(Рис. 8).
 |
 |
||||
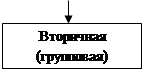 |
|||||
Рис. 8. Классификация упаковки по применению
Первичная (индивидуальная) упаковка предназначается для создания необходимых условий, обеспечивающих длительную сохранность заключенной в ней продукции.
К первичной упаковке относятся: флаконы и банки из стекла с винтовой горловиной, флаконы и банки из дрота, банки из стекла с треугольным венчиком, бутылки для крови и кровезаменителей, полимерные емкости, капсулы, тубы алюминиевые, шприц-тюбики разового применения, аэрозольные баллоны с защитным полиэтиленовым или полимерным покрытием на основе поливинилхлорида, пакеты из полимерных материалов или бумаги, пробирки из дрота, металла или пластмассы, контурная тара, завертка брикета (лекарственное растительное сырье) в этикетку-бандероль.
Уделяется большое внимание наличию необходимых потребительских свойств упаковки:
· транспортабельность упаковки (при ношении, перевозке);
· наличие информации о хранении и приеме ЛС;
· приятный внешний вид;
· соответствующие размеры, обеспечивающие удобство пользования и комплектность;
· простота уничтожения использованной упаковки или возможность повторного использования упаковки как по прямому назначению, так и в других целях.
Следует сказать о специальных требованиях, таких как:
· контроль первого вскрытия упаковки;
· особое размещение ЛС с возможностью многократного использования без нарушения герметичности, стерильности;
· контроль за использованием ЛС.
Выбор того или иного вида упаковки в первую очередь диктуется свойствами лекарственного препарата, определяющими характер используемых упаковочных материалов, вид и конструктивные признаки упаковки, исходя из максимального удовлетворения потребительских требований и соблюдения интересов производства.
Вторичная (групповая) упаковка объединяет некоторое количество первичных упаковок и предназначается для обеспечения их сохранности.
Основными функциями вторичной упаковки являются:
1) сохранность первичной упаковки от атмосферных воздействий;
2) возможность наиболее простого, удобного учета и контроля продукции;
3) удовлетворение потребностей потребителей в информации о ЛС.
Виды вторичной упаковки: картонная пачка с инструкцией и наклеенной этикеткой, упаковка из полимерной пленки и фольги, банка стеклянная, пакеты или мешки из крафт-бумаги, мешки пленочные из полимерных материалов, обертка бумажная с бандеролью и этикеткой (для предметов санитарии и гигиены).
Третичная или транспортная упаковка предназначена для поставки продукции до мест распределения и реализации. Как правило, до потребителя она не доходит.
Согласно существующим требованиям, транспортная упаковка должна защищать ЛС от воздействия осадков и пыли, солнечного облучения, механических повреждений.
Виды транспортной упаковки: короб из гофрированного картона, ящики деревянные, контейнер, мешки из полимерных материалов, мешки из крафт-бумаги, тканевые.
В практике могут возникать различные варианты, когда используется несколько вторичных упаковок или отсутствует транспортная упаковка, но в большинстве случаев указанная классификация вполне приемлема.
3.2.Требования к упаковке лекарственных средств
Современные ЛП отличает огромное количество различных вариантов и форм упаковки. Несмотря на такое разнообразие можно сформулировать основные требования, которые должны выполняться независимо от формы используемой упаковки.
Эти требования можно условно разделить на четыре типа:
1. Конструктивные требования к первичной упаковке.
2. Требования к материалам.
3. Специфические требования, зависящие от типа препарата, конструкции упаковки и технологии изготовления.
4. Общие требования к упаковке.
1. Конструкция первичной упаковки должна обеспечивать:
· защиту ЛП от воздействия неблагоприятных воздействий внешней среды;
· предохранять от механических воздействий;
· обеспечить герметичность и стабильность;
· защиту от микробного загрязнения;
· дозированное или поштучное извлечение ЛП;
· эстетичный внешний вид и удобство использования;
· элементы конструкции должны быть стандартизированы, не должно быть отклонений от геометрических размеров;
· элементы первичной упаковки должны быть сконструированы с возможностью их автоматической обработки и герметичного соединения на автоматическом оборудовании.
2. Материалы первичной упаковки не должны содержать:
· тяжелых металлов, мышьяка, других вредных примесей, в количествах, превышающих нормативы;
· красителей, не разрешенных к применению;
· канцерогенных и токсичных компонентов;
· постороннего запаха;
· микробной обсемененности выше установленных норм;
Не допускается:
· повреждения защитных покрытий;
· наличия механических загрязнений;
· материалы не должны быть хрупкими и должны выдерживать термическую и механическую обработку, обработку дезинфицирующими растворами;
· материалы должны быть нейтральными и не вступать во взаимодействие с компонентами ЛП.
3. Специфические требования к упаковке определяются в основном типом лекарственного препарата и технологическим процессом его изготовления. Например, при хранении ряда препаратов не допускается воздействие на них прямого солнечного света, поэтому упаковка должна быть непрозрачной или, например, для стеклянных флаконов выполнена из оранжевого стекла. Для инъекционных растворов, глазных капель, наоборот, упаковка должна быть максимально прозрачна для возможности контроля микро загрязнений.
4. Общие требования к упаковке:
· четкость напечатанных на упаковке текстов;
· краткая аннотация или инструкция по применению;
· цветное оформление;
· отсутствие вспомогательных средств для вскрытия упаковки;
· по возможности наличие контроля первого вскрытия;
· безопасность в обращении, отсутствие острых углов и краев.
3.3.Понятие маркировки. Производственные маркировочные надписи
Маркировка — это одно из средств товарной информации, которое представляет собой текст, условные обозначения или рисунок, нанесенные на упаковку и (или) товар и предназначенные для идентификации товара или отдельных его свойств, доведения до потребителя информации об изготовителях, качественных и количественных характеристиках товара.
В зависимости от места нанесения различают производственную и торговую маркировку.
Особенно жесткие требования предъявляются к производственной маркировке фармацевтической продукции, которая регламентируется Федеральными законами “О стандартизации” и “Об обращении лекарственных средств”, Методическими указаниями Министерства здравоохранения и социального развития Российской Федерации 9467-015-05749470-98 “Графическое оформление лекарственных средств. Общие требования”.
|
К производственным маркировочным надписям можно отнести: серию, лот, дату производства. Серия (по-английски Batch) - это определенное количество продукта, выработанное без изменения условий из определенного количества сырья без остановки производства (как бы из одного «замеса»).
Партия (по-английски Lot) - это количество продукции (возможно, различных серий) одномоментно выставляемого для продажи (или отправляемое в адрес покупателя). Номера серий могут учитывать дату производства. Примеры обозначения серий на упаковках: Серия № 601198 (последние четыре цифры обозначают месяц и год производства); Серия № 034100 (серия не учитывает дату производства) Серия № 9710239 (первые четыре цифры обозначают год и месяц производства); Серия: 146732/141372. Слово «серия» не всегда присутствует на отечественной упаковке. Иногда штампом просто наносится пяти-, шести- или семизначная цифра. Иностранный вариант обозначения серии: В. No 020693 (учитывает месяц и год производства); Ch.- В. 210053 Charge - Nr.: 1530799 (учитывает месяц и год производства) No. 0301192 или В 0615. Иногда вместо серии на упаковке импортной продукции указывается контрольный номер, например: «Контрольный N 023079» или «O.N. 1109/56». Лот или номер партии указывается на импортных упаковках. Примеры: LOT#0471 или LOT No. 67.
Для обозначения сроков годности могут использоваться различные варианты. Срок годности исчисляется с момента производства. Поэтому в маркировочных надписях чаще всего присутствует информация о дате производства (она может быть включена в номер серии) и (или) указывается дата истечения срока годности. Другой вариант: указана дата производства и количество дней, месяцев или лет, в течение которых продукция пригодна к употреблению. Например: «Дата выпуска V — 2010 год. Срок годности 5 лет». Сроки годности часто обозначаются надписью «Годен до», например: Годен до: 10 10 Годен до: 1 12 2007 Годность до: 11 2014 Дата истечения срока годности импортных лекарственных средств и БАД обозначается с использованием слова Expiry (англ. «истекать»).
Примеры обозначений срока годности: ЕХР 9/14; EXP SEP 14 и т.д. На этикетках к минеральным водам сроки изготовления (розлива) указываются просечками на специальной линейной шкале «месяцы—годы», расположенной на одной из боковых полос этикетки, а словами пишется в течение какого срока годен продукт (обычно 12 месяцев).
Дата производства может быть включена в номер серии, а может указываться отдельно. В английской транскрипции часто используется сокращенный вариант слова «Manufactured « — «Mfd» — произведено (иногда «Mfg»). Пример. Дата произв.: 08/11 MFD 7/10 008 MFD 1011. |
К производственным надписям так же обязательно относят Регистрационный номер.
Например, в соответствии с МУ по графическому оформлению лекарственных средств регистрационный номер лекарственного препарата указывается буквой “Р” и арабскими цифрами: первые две цифры обозначают год издания приказа Министерства здравоохранения и социального развития Российской федерации, согласно которому разрешены промышленный выпуск и применение данного лекарственного средства в Российской федерации; следующая группа цифр — номер приказа и последняя группа цифр — номер пункта в приказе.
Указанные группы цифр разделяются точками или косой чертой (Р 98.211.14 или Р 98/211/11). Приведенный регистрационный номер расшифровывается следующим образом: лекарственный препарат зарегистрирован Приказом Министерства здравоохранения и социального развития Российской Федерации № 211 от 1998 г., п. 14.
3.4. Маркировка лекарственных средств
В соответствии с Федеральным законом “О защите прав потребителя” текст производственной маркировки должен быть на русском языке и доступным для чтения (изготовитель и (или) продавец обязаны сделать перевод текста маркировки на русский язык и сообщить его потребителю любым доступным способом).
Лекарственные препараты, за исключением лекарственных препаратов, изготовленных аптечными организациями, ветеринарными аптечными организациями, индивидуальными предпринимателями, имеющими лицензию на фармацевтическую деятельность, должны поступать в обращение, если:
1) на их первичной упаковке хорошо читаемым шрифтом на русском языке указаны наименование лекарственного препарата (международное непатентованное, или химическое, или торговое наименование), номер серии, дата выпуска (для иммунобиологических лекарственных препаратов), срок годности, доза и форма выпуска, объем и количество доз (для иммунобиологических лекарственных препаратов);
2) на их вторичной (потребительской) упаковке хорошо читаемым шрифтом на русском языке указаны наименование лекарственного препарата (международное непатентованное или химическое и торговое наименования), наименование производителя лекарственного препарата, номер серии, дата выпуска (для иммунобиологических лекарственных препаратов), номер регистрационного удостоверения, срок годности, способ применения, доза и количество доз в упаковке, форма выпуска, условия отпуска, условия хранения, меры предосторожности при применении лекарственного препарата, предупредительные надписи.
Лекарственные средства в качестве сывороток должны поступать в обращение с указанием животного, из крови, плазмы крови, органов и тканей которого они получены.
На вторичную (потребительскую) упаковку лекарственных средств, полученных из крови, плазмы крови, органов и тканей человека, должна наноситься надпись: "Антитела к ВИЧ-1, ВИЧ-2, к вирусу гепатита С и поверхностный антиген вируса гепатита В отсутствуют".
На вторичную (потребительскую) упаковку гомеопатических лекарственных препаратов должна наноситься надпись: "Гомеопатический".
На вторичную (потребительскую) упаковку лекарственных растительных препаратов должна наноситься надпись: "Продукция прошла радиационный контроль".
Упаковка лекарственных средств, предназначенных исключительно для экспорта, маркируется в соответствии с требованиями страны-импортера.
На первичную упаковку и вторичную (потребительскую) упаковку лекарственных средств для ветеринарного применения должна наноситься надпись: "Для ветеринарного применения".
На вторичную (потребительскую) упаковку лекарственного препарата наносится штриховой код.
Данные условия регламентирует ст. 46 «Маркировка лекарственных средств» ФЗ № 61 «Об обращении лекарственных средствах» и называется «Маркировка и оформление лекарственных средств».
Этикетка содержит марочное название, марочный знак, рекламные материалы, инструкции для использования; вкладыш, дает детальные указания по применению, мерам предосторожности и т.д.
Среди всех функций маркировки (информационной, идентифицирующей, мотивационной, эмоциональной) самой важной является идентифицирующая, так как именно с идентификации товара начинается его экспертная оценка. Установление соответствия характеристик товара, указанных на маркировке, в товарно-сопроводительных документах или иных средствах информации, предъявляемым требованиям, определяется с помощью идентификации.
3.5. Понятие и классификация товарных знаков
Товарный знак — это любое название, символ, рисунок или их комбинация, используемые для обозначения товаров компании, отличающие их от товаров конкурентов. Право на товарный знак охраняется законом. Регистрация товарного знака действует в течение 10 лет.
|
Классификация товарных знаков осуществляется по различным признакам, положенным в основу: по объектам, информацию о которых они содержат; по форме представления информации; по виду собственности. Товарные знаки делятся: 1. По объектам товарной информации: - фирменные (обыкновенные и престижные); - ассортиментные (видовые и марочные). 2. По форме представления информации: - словесные; - буквенные; - цифровые; - объемные; - изобразительные; - световые; - комбинированные. 3. По виду собственности владельца: - индивидуальные; - коллективные. Фирменные товарные знаки - знаки, которые предназначены для идентификации изготовителя товаров или услуг. Товарные
знаки фирм могут состоять из следующих элементов: 1. слов, букв, группы слов или букв, которые могут быть произнесены и образуют фирменное имя; 2. символов, рисунков, отличительных цветов и обозначений, которые и образуют фирменный знак; 3. знака защиты - R или С. Все данные элементы могут комбинироваться или совмещаться в виде торгового знака. Ассортиментные (именные) товарные знаки - знаки, предназначенные для идентификации ассортиментной принадлежности. Делятся на марочные и видовые. Видовые товарные знаки идентифицируют товар по виду. Видовым товарным знаком может служить словесная информация о названии товара, его изображение или условное буквенное обозначение определенного вида продукта. Чаще всего видовые знаки применяются в комбинации с марочными. Марочный товарный знак (торговая марка) - обозначения, предназначенные для идентификации и дифференциации товаров (услуг) одних продавцов от аналогичных товаров (услуг) конкурентов. Товарные марки появились в Средние века, когда гильдии ремесленников и торговцев требовали, чтобы каждый производитель помечал свои товары так, чтобы можно было контролировать объем производства и выявлять изготовителей низкокачественной продукции. Первыми пропагандистами товарных марок, например в США, были изготовители патентованных медицинских средств. По-настоящему большое распространение марочные названия получили с появлением общенациональных фирм и общенациональных средств рекламы. Каждая фирма принимает собственное решение относительно того, будет ли она присваивать своему товару марочное название или нет. Если принимается положительное решение, то далее обсуждается вопрос о том, каким будет это марочное название — индивидуальным, единым для всех товаров фирмы, коллективным для семейств товаров или включающим название самой фирмы. Марочный товарный знак может включать следующие элементы: 1. Марочное название (имя) — часть марки, которую можно произнести (слова, буквы, цифры или их комбинации); 2. Марочный знак (эмблему) — часть марки, которую можно опознать, но невозможно произнести (например, символ, изображение, отличительную окраску и специфическое шрифтовое оформление). Марочное название в сочетании с марочным знаком часто составляют логотип, идентифицирующий продукцию определенной фирмы. ™ — знак, защищающий исключительные права продавца на пользование марочным названием и (или) марочным знаком. В ряде случаев защитным знаком является ®. В таком случае в маркировочных надписях, как правило, делается сноска о том, что данное название является зарегистрированной торговой маркой соответствующей фирмы. |
3.6. Знаки соответствия или качества
|
Знак соответствия (в области сертификации) удостоверяет, что данная продукция соответствует конкретному стандарту или другому нормативному документу. В зависимости от сферы применения различают национальные и транснациональные знаки соответствия. Национальный знак соответствия - знак, подтверждающий соответствие требованиям, установленным национальными стандартами или другими нормативными документами. Он разрабатывается, утверждается и регистрируется национальным органом по стандартизации и сертификации. Знак соответствия разрешается использовать для маркирования только сертифицированной продукции. Заявители (изготовители, продавцы) любой страны могут маркировать свою продукцию национальным знаком соответствия при наличии сертификата (выданного одним из национальных органов по сертификации или при наличии соглашений о взаимном признании результатов сертификации), а также после получения лицензии на применение знака соответствия. Национальные знаки соответствия могут быть общими для всех видов продукции или групповыми, подтверждающими соответствие определенной группы (или однородных групп) продукции. В России утвержден только общий знак соответствия системы ГОСТ Р. Наличие этого знака означает, что данная продукция соответствует государственным стандартам Российской Федерации. Через аптечную сеть реализуется достаточно большое количество упаковок, имеющих российский национальный знак соответствия. Знаки соответствия стандартам других стран можно встретить на упаковках с продукцией медицинского назначения импортного производства. Групповой национальный знак соответствия, предназначенный для маркирования российской продукции медицинского назначения, изображен слева.
Транснациональные (региональные) знаки соответствия - знаки, подтверждающие соответствие требованиям, установленным региональными стандартами. Они применяются в странах определенного региона на основе гармонизированных стандартов и взаимного признания результатов сертификации. Наряду со знаками соответствия в ряде стран применяются и знаки качества. В отличие от первых знаки качества могут присваиваться не только органами по сертификации, но и другими организациями, не входящими в национальную систему сертификации. Знаки качества могут: - подразумевать гарантии длительного и (или) успешного использования продукта; - сообщать о рекомендациях (или одобрении) компетентных органов по использованию данного продукта; - сообщать о тестировании или проведении
сертификации данного продукта по каким-либо показателям авторитетным
органом; - сообщать название фирмы и год ее основания, который свидетельствует о традиционно высоком качестве продукта за весь период существования на рынке. |
ГЛАВА 4. ОРГАНИЗАЦИЯ ХРАНЕНИЯ ЛЕКАРСТВЕННЫХ СРЕДСТВ И ДРУГИХ ТОВАРОВ АПТЕЧНОГО АССОРТИМЕНТА
4.1.Общие требования к устройству и эксплуатации помещений хранения лекарственных средств
Устройство, состав, размеры площадей, эксплуатация и оборудование помещений для хранения лекарственных средств должны обеспечивать их сохранность.
В помещениях для хранения лекарственных средств должны поддерживаться определенные температура и влажность воздуха, позволяющие обеспечить хранение лекарственных средств в соответствии с указанными на первичной и вторичной (потребительской) упаковке требованиями производителей лекарственных средств.
Помещения для хранения лекарственных средств должны быть оборудованы кондиционерами и другим оборудованием, позволяющим обеспечить хранение лекарственных средств в соответствии с указанными на первичной и вторичной (потребительской) упаковке требованиями производителей лекарственных средств, либо помещения рекомендуется оборудовать форточками, фрамугами, вторыми решетчатыми дверьми.
Помещения для хранения лекарственных средств должны быть обеспечены стеллажами, шкафами, поддонами, подтоварниками.
Отделка помещений для хранения лекарственных средств (внутренние поверхности стен, потолков) должна быть гладкой и допускать возможность проведения влажной уборки.
4.2.Общие требования к помещениям для хранения лекарственных средств и организации их хранения
Помещения для хранения лекарственных средств должны быть оснащены приборами для регистрации параметров воздуха (термометрами, гигрометрами (электронными гигрометрами) или психрометрами). Измерительные части этих приборов должны размещаться на расстоянии не менее 3 м от дверей, окон и отопительных приборов. Приборы и (или) части приборов, с которых производится визуальное считывание показаний, должны располагаться в доступном для персонала месте на высоте 1,5 - 1,7 м от пола.
Показания этих приборов должны ежедневно регистрироваться в специальном журнале (карте) регистрации на бумажном носителе или в электронном виде с архивацией (для электронных гигрометров), который ведется ответственным лицом. Журнал (карта) регистрации хранится в течение одного года, не считая текущего. Контролирующие приборы должны быть сертифицированы, калиброваны и подвергаться поверке в установленном порядке.
В помещениях для хранения лекарственные средства размещают в соответствии с требованиями нормативной документации, указанной на упаковке лекарственного препарата, с учетом:
физико-химических свойств лекарственных средств;
фармакологических групп (для аптечных и медицинских организаций);
способа применения (внутреннее, наружное);
агрегатного состояния фармацевтических субстанций (жидкие, сыпучие, газообразные).
При размещении лекарственных средств допускается использование компьютерных технологий (по алфавитному принципу, по кодам).
Отдельно, в технически укрепленных помещениях, соответствующих требованиям Федерального закона от 8 января 1998 г. N 3-ФЗ "О наркотических средствах и психотропных веществах" хранятся:
- наркотические и психотропные лекарственные средства;
- сильнодействующие и ядовитые лекарственные средства, находящиеся под контролем в соответствии с международными правовыми нормами.
Стеллажи (шкафы) для хранения лекарственных средств в помещениях для хранения лекарственных средств должны быть установлены таким образом, чтобы обеспечить доступ к лекарственным средствам, свободный проход персонала и, при необходимости, погрузочных устройств, а также доступность стеллажей, стен, пола для уборки.
Стеллажи, шкафы, полки, предназначенные для хранения лекарственных средств, должны быть идентифицированы (промаркированы).
Хранящиеся лекарственные средства должны быть также идентифицированы с помощью стеллажной карты, содержащей информацию о хранящемся лекарственном средстве (наименование, форма выпуска и дозировка, номер серии, срок годности, производитель лекарственного средства). При использовании компьютерных технологий допускается идентификация при помощи кодов и электронных устройств.
В организациях и у индивидуальных предпринимателей необходимо вести учет лекарственных средств с ограниченным сроком годности на бумажном носителе или в электронном виде с архивацией. Контроль за своевременной реализацией лекарственных средств с ограниченным сроком годности должен осуществляться с использованием компьютерных технологий, стеллажных карт с указанием наименования лекарственного средства, серии, срока годности либо журналов учета сроков годности. Порядок ведения учета указанных лекарственных средств устанавливается руководителем организации или индивидуальным предпринимателем.
При выявлении лекарственных средств с истекшим сроком годности они должны храниться отдельно от других групп лекарственных средств в специально выделенной и обозначенной (карантинной) зоне.
4.3. Особенности хранения отдельных групп лекарственных средств в зависимости от физических и физико-химических свойств, воздействия на них различных
факторов внешней среды.
4.3.1. Хранение лекарственных средств, требующих защиты от действия света
Лекарственные средства, требующие защиты от действия света, хранятся в помещениях или специально оборудованных местах, обеспечивающих защиту от естественного и искусственного освещения.
Фармацевтические субстанции, требующие защиты от действия света, следует хранить в таре из светозащитных материалов, в темном помещении или шкафах.
Для хранения особо чувствительных к свету фармацевтических субстанций (нитрат серебра, Прозерпин) стеклянную тару оклеивают черной светонепроницаемой бумагой.
Лекарственные препараты для медицинского применения, требующие защиты от действия света, упакованные в первичную и вторичную (потребительскую) упаковку, следует хранить в шкафах или на стеллажах при условии принятия мер для предотвращения попадания на указанные лекарственные препараты прямого солнечного света или иного яркого направленного света (использование светоотражающей пленки, жалюзи, козырьков и др.).
4.3.2. Хранение лекарственных средств, требующих защиты от воздействия влаги
Фармацевтические субстанции, требующие защиты от воздействия влаги, следует хранить в прохладном месте при температуре до +15 град. C (далее - прохладное место), в плотно укупоренной таре из материалов, непроницаемых для паров воды (стекла, металла, алюминиевой фольги, толстостенной пластмассовой таре) или в первичной и вторичной (потребительской) упаковке производителя.
Фармацевтические субстанции с выраженными гигроскопическими свойствами следует хранить в стеклянной таре с герметичной укупоркой, залитой сверху парафином.
Во избежание порчи и потери качества следует организовать хранение лекарственных средств в соответствии с требованиями, нанесенными в виде предупреждающих надписей на вторичной (потребительской) упаковке лекарственного средства.
4.3.3. Хранение лекарственных средств, требующих защиты от улетучивания и высыхания
Фармацевтические субстанции, требующие защиты от улетучивания и высыхания (собственно летучие лекарственные средства; лекарственные средства, содержащие летучий растворитель (спиртовые настойки, жидкие спиртовые концентраты, густые экстракты); растворы и смеси летучих веществ (эфирные масла, растворы аммиака, формальдегида, хлористого водорода свыше 13%, карболовой кислоты, этиловый спирт различной концентрации и др.); лекарственное растительное сырье, содержащее эфирные масла; лекарственные средства, содержащие кристаллизационную воду, - кристаллогидраты; лекарственные средства, разлагающиеся с образованием летучих продуктов (йодоформ, перекись водорода, гидрокарбонат натрия); лекарственные средства с определенным нижним пределом влагосодержания (сульфат магния, сульфат натрия)), следует хранить в прохладном месте, в герметически укупоренной таре из непроницаемых для улетучивающихся веществ материалов (стекла, металла, алюминиевой фольги) или в первичной и вторичной (потребительской) упаковке производителя. Применение полимерной тары, упаковки и укупорки допускается в соответствии с требованиями государственной фармакопеи и нормативной документации.
4.3.4. Хранение лекарственных средств, требующих защиты от воздействия повышенной температуры
Хранение лекарственных средств, требующих защиты от воздействия повышенной температуры (термолабильные лекарственные средства), организации и индивидуальные предприниматели должны осуществлять в соответствии с температурным режимом, указанным на первичной и вторичной (потребительской) упаковке лекарственного средства в соответствии с требованиями нормативной документации.
4.3.5. Хранение лекарственных средств, требующих защиты от воздействия пониженной температуры
Хранение лекарственных средств, требующих защиты от воздействия пониженной температуры (лекарственные средства, физико-химическое состояние которых после замерзания изменяется и при последующем согревании до комнатной температуры не восстанавливается (40% раствор формальдегида, растворы инсулина)), организации и индивидуальные предприниматели должны осуществлять в соответствии с температурным режимом, указанным на первичной и вторичной (потребительской) упаковке лекарственного средства в соответствии с требованиями нормативной документации.
Замерзание препаратов инсулина не допускается.
4.3.6. Хранение лекарственных средств, требующих защиты от воздействия газов, содержащихся в окружающей среде
Фармацевтические субстанции, требующие защиты от воздействия газов (вещества, реагирующие с кислородом воздуха: морфин и его производные; серосодержащие соединения, ферменты и органопрепараты; вещества, реагирующие с углекислым газом воздуха: соли щелочных металлов и слабых органических кислот (барбитал натрий, гексенал), эуфиллин, окись и перекись магния, едкий натрий и калий), следует хранить в герметически укупоренной таре из материалов, непроницаемых для газов, по возможности заполненной доверху.
4.3.7. Хранение пахучих и красящих лекарственных средств
Пахучие лекарственные средства (фармацевтические субстанции как летучие, так и практически нелетучие, но обладающие сильным запахом) следует хранить в герметически закрытой таре, непроницаемой для запаха.
Красящие лекарственные средства (фармацевтические субстанции, которые оставляют окрашенный след, не смываемый обычной санитарно-гигиенической обработкой, на таре, укупорочных средствах, оборудовании и инвентаре (бриллиантовый зеленый, метиленовый синий, индигокармин)) следует хранить в специальном шкафу в плотно укупоренной таре.
Для работы с красящими лекарственными средствами для каждого наименования необходимо выделять специальные весы, ступку, шпатель и другой необходимый инвентарь.
4.3.8. Хранение дезинфицирующих лекарственных средств
Дезинфицирующие лекарственные средства следует хранить в герметично укупоренной таре в изолированном помещении вдали от помещений хранения пластмассовых, резиновых и металлических изделий и помещений получения дистиллированной воды.
4.3.9. Хранение лекарственного растительного сырья
Нерасфасованное лекарственное растительное сырье должно храниться в сухом (не более 50% влажности), хорошо проветриваемом помещении в плотно закрытой таре.
Нерасфасованное лекарственное растительное сырье, содержащее эфирные масла, хранится изолированно в хорошо укупоренной таре.
Нерасфасованное лекарственное растительное сырье должно подвергаться периодическому контролю в соответствии с требованиями государственной фармакопеи. Трава, корни, корневища, семена, плоды, утратившие нормальную окраску, запах и требуемое количество действующих веществ, а также пораженные плесенью, амбарными вредителями, бракуют.
Хранение лекарственного растительного сырья, содержащего сердечные гликозиды, осуществляется с соблюдением требований государственной фармакопеи, в частности, требования о повторном контроле на биологическую активность.
Нерасфасованное лекарственное растительное сырье, включенное в списки сильнодействующих и ядовитых веществ, утвержденные Постановлением Правительства Российской Федерации от 29 декабря 2007 г. N 964 "Об утверждении списков сильнодействующих и ядовитых веществ» хранится в отдельном помещении или в отдельном шкафу под замком.
Расфасованное лекарственное растительное сырье хранится на стеллажах или в шкафах.
4.3.10. Хранение медицинских пиявок
Хранение медицинских пиявок осуществляется в светлом помещении без запаха лекарств, для которого устанавливается постоянный температурный режим.
Содержание пиявок осуществляется в установленном порядке.
4.4. Требования к помещениям для хранения огнеопасных и взрывоопасных лекарственных средств и организации их хранения
Помещения для хранения огнеопасных и взрывоопасных лекарственных средств должны полностью соответствовать действующим нормативным документам.
Помещения для хранения лекарственных средств в организациях оптовой торговли лекарственными средствами и у производителей лекарственных средств разбиваются на отдельные помещения с пределом огнестойкости строительных конструкций не менее 1 часа с целью обеспечения хранения огнеопасных и взрывоопасных лекарственных средств по принципу однородности в соответствии с их физико-химическими, пожароопасными свойствами и характером упаковки.
Необходимое для фасовки и изготовления лекарственных препаратов для медицинского применения на одну рабочую смену количество огнеопасных лекарственных средств допускается содержать в производственных и иных помещениях. Оставшееся количество огнеопасных лекарственных средств по окончании работы в конце смены передается следующей смене или возвращается на место основного хранения.
Полы складских помещений и разгрузочных площадок должны иметь твердое, ровное покрытие. Запрещается применять доски и железные листы для выравнивания полов. Полы должны обеспечивать удобное и безопасное передвижение людей, грузов и транспортных средств, обладать достаточной прочностью и выдерживать нагрузки от хранимых материалов, обеспечивать простоту и легкость уборки складского помещения.
Складские помещения для хранения огнеопасных и взрывоопасных лекарственных средств должны быть оборудованы несгораемыми и устойчивыми стеллажами и поддонами, рассчитанными на соответствующую нагрузку. Стеллажи устанавливаются на расстоянии 0,25 м от пола и стен, ширина стеллажей не должна превышать 1 м. Продольные проходы между стеллажами должны быть не менее 1,35 м.
В аптечных организациях и у индивидуальных предпринимателей выделяются изолированные помещения, оборудуемые средствами автоматической пожарной защиты и сигнализацией, для хранения огнеопасных фармацевтических субстанций и взрывоопасных лекарственных средств.
В аптечных организациях и у индивидуальных предпринимателей допускается хранение фармацевтических субстанций, обладающих легковоспламеняющимися и горючими свойствами, в объеме до 10 кг вне помещений для хранения огнеопасных фармацевтических субстанций и взрывоопасных лекарственных средств во встроенных несгораемых шкафах. Шкафы должны быть удалены от тепловыводящих поверхностей и проходов, с дверьми шириной не менее 0,7 м и высотой не менее 1,2 м. К ним должен быть организован свободный доступ.
Допускается хранение взрывоопасных лекарственных препаратов для медицинского применения (во вторичной (потребительской) упаковке) для использования на одну рабочую смену в металлических шкафах вне помещений для хранения огнеопасных фармацевтических субстанций и взрывоопасных лекарственных средств.
Количество огнеопасных фармацевтических субстанций, допустимое для хранения в помещениях для хранения огнеопасных фармацевтических субстанций и взрывоопасных лекарственных средств, расположенных в зданиях другого назначения, не должно превышать 100 кг в нерасфасованном виде.
Помещения для хранения огнеопасных фармацевтических субстанций и взрывоопасных лекарственных средств, используемые для хранения легковоспламеняющихся фармацевтических субстанций в количестве свыше 100 кг, должны находиться в отдельно стоящем здании, а само хранение должно осуществляться в стеклянной или металлической таре изолированно от помещений для хранения других групп огнеопасных фармацевтических субстанций.
В помещения для хранения огнеопасных фармацевтических субстанций и взрывоопасных лекарственных средств запрещается входить с открытыми источниками огня.
4.4.1. Хранение огнеопасных лекарственных средств
Хранение огнеопасных лекарственных средств (лекарственные средства, обладающие легковоспламеняющимися свойствами (спирт и спиртовые растворы, спиртовые и эфирные настойки, спиртовые и эфирные экстракты, эфир, скипидар, молочная кислота, хлорэтил, коллодий, клеол, жидкость Новикова, органические масла); лекарственные средства, обладающие легкогорючими свойствами (сера, глицерин, растительные масла, нерасфасованное лекарственное растительное сырье)) должно осуществляться отдельно от других лекарственных средств.
Легковоспламеняющиеся лекарственные средства хранят в плотно укупоренной прочной, стеклянной или металлической таре, чтобы предупредить испарение жидкостей из сосудов.
Бутыли, баллоны и другие крупные емкости с легковоспламеняющимися и легкогорючими лекарственными средствами должны храниться на полках стеллажей в один ряд по высоте. Запрещается их хранение в несколько рядов по высоте с использованием различных прокладочных материалов.
Не допускается хранение указанных лекарственных средств у отопительных приборов. Расстояние от стеллажа или штабеля до нагревательного элемента должно быть не менее 1 м.
Хранение бутылей с легковоспламеняющимися и легкогорючими фармацевтическими субстанциями должно осуществляться в таре, предохраняющей от ударов, или в баллоно-опрокидывателях в один ряд.
На рабочих местах производственных помещений, выделяемых в аптечных организациях и индивидуальными предпринимателями, легковоспламеняющиеся и легкогорючие лекарственные средства могут храниться в количествах, не превышающих сменную потребность. При этом емкости, в которых они хранятся, должны быть плотно закрыты.
Не допускается хранение легковоспламеняющихся и легкогорючих лекарственных средств в полностью заполненной таре. Степень заполнения должна быть не более 90% объема. Спирты в больших количествах хранятся в металлических емкостях, заполняемых не более чем на 75% объема.
Не допускается совместное хранение легковоспламеняющихся лекарственных средств с минеральными кислотами (особенно серной и азотной кислотами), сжатыми и сжиженными газами, легкогорючими веществами (растительными маслами, серой, перевязочным материалом), щелочами, а также с неорганическими солями, дающими с органическими веществами взрывоопасные смеси (калия хлорат, калия перманганат, калия хромат и др.).
Эфир медицинский и эфир для наркоза хранят в промышленной упаковке, в прохладном, защищенном от света месте, вдали от огня и нагревательных приборов.
4.4.2. Хранение взрывоопасных лекарственных средств
При хранении взрывоопасных лекарственных средств (лекарственные средства, обладающие взрывчатыми свойствами (нитроглицерин); лекарственные средства, обладающие взрывоопасными свойствами (калия перманганат, серебра нитрат)) следует принимать меры против загрязнения их пылью.
Емкости с взрывоопасными лекарственными средствами (штангласы, жестяные барабаны, склянки и др.) необходимо плотно закрывать во избежание попадания паров этих средств в воздух.
Хранение нерасфасованного калия перманганата допускается в специальном отсеке складских помещений (где он хранится в жестяных барабанах), в штангласах с притертыми пробками отдельно от других органических веществ - в аптечных организациях и у индивидуальных предпринимателей.
Нерасфасованный раствор нитроглицерина хранится в небольших хорошо укупоренных склянках или металлических сосудах в прохладном, защищенном от света месте, с соблюдением мер предосторожности от огня. Передвигать посуду с нитроглицерином и отвешивать этот препарат следует в условиях, исключающих пролив и испарение нитроглицерина, а также попадание его на кожу.
Запрещается хранение взрывоопасных лекарственных средств с кислотами и щелочами.
4.5. Правила хранения медицинских изделий
Требования к хранению медицинских изделий устанавливаются производителем медицинских изделий.
Хранение медицинских изделий осуществляется производителями, организациями оптовой торговли медицинских изделий, аптечными организациями, индивидуальными предпринимателями, имеющими лицензию на медицинскую деятельность, медицинскими организациями и иными организациями, осуществляющими обращение медицинских изделий.
В аптечных учреждениях хранение медицинских изделий осуществляется по группам:
- резиновые изделия;
- изделия из пластмасс;
- перевязочные средства и вспомогательные материалы;
- иные медицинские изделия.
Резиновые изделия
Для наилучшего сохранения резиновых изделий в помещениях хранения необходимо создать:
- защиту от света, особенно прямых солнечных лучей, высокой (более 20 град.С) и низкой (ниже 0 град.С) температуры воздуха; текучего воздуха (сквозняков, механической вентиляции); механических повреждений (сдавливания, сгибания, скручивания, вытягивания и т.п.);
- для предупреждения высыхания, деформации и потери их эластичности, относительную влажность не менее 65%;
- изоляцию от воздействия агрессивных веществ (йод, хлороформ, хлористый аммоний, лизол, формалин, кислоты, органические растворители, смазочных масел и щелочей, хлорамин Б, нафталин);
- условия хранения вдали от нагревательных приборов (не менее 1 м).
Помещения хранения резиновых изделий должны располагаться не на солнечной стороне, лучше в полуподвальных темных или затемненных помещениях. Для поддержания в сухих помещениях повышенной влажности рекомендуется ставить сосуды с 2% водным раствором карболовой кислоты.
В помещениях, шкафах рекомендуется ставить стеклянные сосуды с углекислым аммонием, способствующим сохранению эластичности резины.
Для хранения резиновых изделий помещения хранения оборудуются шкафами, ящиками, полками, стеллажами, блоками для подвешивания, стойками и другим необходимым инвентарем, с учетом свободного доступа.
При размещении резиновых изделий в помещениях хранения необходимо полностью использовать весь его объем. Это предотвращает вредное влияние избыточного кислорода воздуха. Однако резиновые изделия (кроме пробок) нельзя укладывать в несколько слоев, так как предметы, находящиеся в нижних слоях, сдавливаются и слеживаются.
Шкафы для хранения медицинских резиновых изделий и парафармацевтической продукции этой группы должны иметь плотно закрывающиеся дверцы. Внутри шкафы должны иметь совершенно гладкую поверхность.
Внутреннее устройство шкафов зависит от вида хранящихся в них резиновых изделий. Шкафы, предназначенные для:
- хранения резиновых изделий в лежачем положении (бужи, катетеры, пузыри для льда, перчатки и т.п.), оборудуются выдвижными ящиками с таким расчетом, чтобы в них можно было размещать предметы на всю длину, свободно, не допуская их сгибов, сплющивания, скручивания и т.п.;
- хранения изделий в подвешенном состоянии (жгутов, зондов, ирригаторной трубки), оборудуются вешалками, расположенными под крышкой шкафа. Вешалки должны быть съемными с тем, чтобы их можно было вынимать с подвешенными предметами. Для укрепления вешалок устанавливаются накладки с выемками.
Резиновые изделия размещают в хранилищах по наименованиям и срокам годности. На каждой партии резиновых изделий прикрепляют ярлык с указанием наименования, срока годности.
Особое внимание следует уделить хранению некоторых видов резиновых изделий, требующих специальных условий хранения:
- круги подкладные, грелки резиновые, пузыри для льда рекомендуется хранить слегка надутыми, резиновые трубки хранятся со вставленными на концах пробками;
- съемные резиновые части приборов должны храниться отдельно от частей, сделанных из другого материала;
- изделия, особо чувствительные к атмосферным факторам - эластичные катетеры, бужи, перчатки, напальчники, бинты резиновые и т.п. хранят в плотно закрытых коробках, густо пересыпанных тальком. Резиновые бинты хранят в скатанном виде пересыпанные тальком по всей длине;
- прорезиненную ткань (одностороннюю двухстороннюю) хранят изолированно от веществ, указанных в пункте 8.1.1., в горизонтальном положении в рулонах, подвешенных на специальных стойках. Прорезиненную ткань допускается хранить уложенной не более чем в 5 рядов на гладко отструганных полках стеллажей;
- эластичные лаковые изделия - катетеры, бужи, зонды (на этилцеллюлозном или копаловом лаке), в отличие от резины, хранят в сухом помещении. Признаком старения является некоторое размягчение, клейкость поверхности. Такие изделия бракуются.
Резиновые пробки должны храниться упакованными в соответствии с требованиями действующих технических условий.
Резиновые изделия необходимо периодически осматривать. Предметы, начинающие терять эластичность, должны быть своевременно восстановлены в соответствии с требованиями НТД.
Резиновые перчатки рекомендуется, если они затвердели, слиплись и стали хрупкими, положить не расправляя, на 15 минут в теплый 5% раствор аммиака, затем перчатки разминают и погружают их на 15 минут в теплую (40-50 град. С) воду с 5% глицерина. Перчатки снова становятся эластичными.
Пластмассовые изделия следует хранить в вентилируемом темном помещении, на расстоянии не менее 1 м от отопительных систем. В помещении не должно быть открытого огня, паров летучих веществ. Электроприборы, арматура и выключатели должны быть изготовлены в противопожарном исполнении. В помещении, где хранятся целлофановые, целлулоидные, аминопластовые изделия, следует поддерживать относительную влажность воздуха не выше 65%.
Перевязочные средства хранят в сухом проветриваемом помещении в шкафах, ящиках, на стеллажах и поддонах, которые должны быть выкрашены изнутри светлой масляной краской и содержаться в чистоте. Шкафы, где находятся перевязочные материалы, периодически протирают 0,2% раствора хлорамина или другими разрешенными к применению дезинфекционными средствами.
Стерильный перевязочный материал (бинты, марлевые салфетки, вата) хранятся в заводской упаковке. Запрещается их хранение в первичной вскрытой упаковке.
Нестерильный перевязочный материал (вата, марля) хранят упакованными в плотную бумагу или в тюках (мешках) на стеллажах или поддонах.
Вспомогательный материал (фильтровальная бумага, бумажные капсулы и др.) необходимо хранить в промышленной упаковке в сухих и проветриваемых помещениях в отдельных шкафах в строго гигиенических условиях. После вскрытия промышленной упаковки расфасованное или оставшееся количество вспомогательного материала рекомендуется хранить в полиэтиленовых, бумажных пакетах или мешках из крафт-бумаги.
ГЛАВА 5. СИСТЕМА ГОСУДАРСТВЕННОГО КОНТРОЛЯ КАЧЕСТВА И СЕРТИФИКАЦИИ ЛЕКАРСТВЕННЫХ СРЕДСТВ
5.1. Государственный контроль качества лекарственных средств
Одним из основных документов, составляющих нормативно-правовую базу госконтроля качества ЛС является ФЗ №61 «Об обращении лекарственных средств». Им закреплен приоритет государственного контроля производства, изготовления, качества, эффективности и безопасности лекарственных средств. В законе даются основные понятия в сфере обращения (СО) ЛС:
Лекарственные вещества - вещества или их комбинации, вступающие в контакт с организмом человека или животного, проникающие в органы, ткани организма человека или животного, применяемые для профилактики, диагностики (за исключением веществ или их комбинаций, не контактирующих с организмом человека или животного), лечения заболевания, реабилитации, для сохранения, предотвращения или прерывания беременности и полученные из крови, плазмы крови, из органов, тканей организма человека или животного, растений, минералов методами синтеза или с применением биологических технологий. К лекарственным средствам относятся фармацевтические субстанции и лекарственные.
Лекарственные препараты - лекарственные средства в виде лекарственных форм, применяемые для профилактики, диагностики, лечения заболевания, реабилитации, для сохранения, предотвращения или прерывания беременности.
Качество лекарственного средства - соответствие лекарственного средства требованиям фармакопейной статьи либо в случае ее отсутствия нормативной документации или нормативного документа.
Безопасность лекарственного средства - характеристика лекарственного средства, основанная на сравнительном анализе его эффективности и риска причинения вреда здоровью.
Эффективность лекарственного препарата - характеристика степени положительного влияния лекарственного препарата на течение, продолжительность заболевания или его предотвращение, реабилитацию, на сохранение, предотвращение или прерывание беременности.
Фальсифицированное ЛС - ЛС, сопровождаемое ложной информацией о составе и/или производителе ЛС.
Недоброкачественное лекарственное средство - лекарственное средство, не соответствующее требованиям фармакопейной статьи либо в случае ее отсутствия требованиям нормативной документации или нормативного документа;
Контрафактное лекарственное средство - лекарственное средство, находящееся в обороте с нарушением гражданского законодательства;
Согласно ст. 9, гл. 4 ФЗ «Об обращении ЛС» государственное регулирование отношений, возникающих в сфере обращения ЛС, осуществляется посредством:
· проведения проверок соблюдения субъектами обращения лекарственных средств правил лабораторной и клинической практики при проведении доклинических и клинических исследований лекарственных препаратов для медицинского применения, правил организации производства и контроля качества лекарственных средств, правил оптовой торговли лекарственными средствами, правил отпуска лекарственных препаратов, правил изготовления и отпуска лекарственных препаратов, правил хранения лекарственных средств, правил уничтожения лекарственных средств;
· лицензирования производства лекарственных средств и фармацевтической деятельности, проведения проверок соблюдения лицензионных требований и условий;
· контроля качества лекарственных средств при гражданском обороте;
· выдачи разрешений на ввоз лекарственных средств на территорию Российской Федерации;
· проведения мониторинга безопасности лекарственных препаратов;
· регулирования ценообразования на ЛС.
Государственное регулирование отношений, возникающих в СО ЛС, осуществляется следующими федеральными органами исполнительной власти:
1. органом, в компетенцию которого входят функции по выработке государственной политики и нормативно-правовому регулированию в СО ЛС ( в настоящее время это Министерство здравоохранения и социального развития РФ - МЗ и СР РФ, в составе которого создан Департамент развития фармацевтического рынка и рынка медицинской техники),
2. органом, в компетенцию которого входит осуществление государственного контроля и надзора в СО ЛС (Федеральная служба по надзору в сфере здравоохранения и социального развития - Росздравнадзор),
3. органом, осуществляющим функции по оказанию государственных услуг, управлению государственным имуществом и правоприменительные функции, за исключением функций по контролю и надзору, в СО ЛС (МЗ и СР РФ), а также органами исполнительной власти субъектов Российской Федерации, поскольку в результате проводимой в стране административной реформы произошло разделение функций в сфере обращения ЛС, ранее осуществляемых подразделениями МЗ РФ.
В настоящий момент, для осуществления деятельности по контролю качества ЛС в субъектах РФ действуют:
· территориальные управления Росздравнадзора;
· экспертные организации, заключившие договор о проведении экспертизы качества ЛС с Росзравнадзором, а именно: центры контроля качества ЛС (ЦККЛС) или КАЛ, или филиалы ФГУ НЦ ЭСМП Росзравнадзора, или другие аккредитованные лаборатории;
· органы сертификации ЛС (в федеральных округах РФ).
Во исполнение ФЗ «Об обращении ЛС» принят приказ МЗ и СР РФ № 734 от 30.10.2006 «Об утверждении административного регламента Федеральной службы по надзору в сфере здравоохранения и социального развития по исполнению государственной функции по организации проведения экспертизы качества, эффективности и безопасности лекарственных средств», согласно которому государственный контроль качества ЛС осуществляется в виде:
· проведения экспертизы качества, эффективности и безопасности лекарственных средств при государственной регистрации;
· осуществления сбора и анализа информации о побочных эффектах применения лекарственных средств;
· осуществления сбора и анализа информации о качестве лекарственных средств;
· предварительного контроля качества ЛС;
· выборочного контроля качества ЛС;
· повторного выборочного контроля качества ЛС.
5.2. Виды государственного контроля
1. Предварительный контроль качества ЛС.
Предварительному контролю подлежат ЛС:
· впервые производимые предприятием;
· впервые ввозимые на территорию РФ;
· выпускаемые по измененной технологии;
· выпускаемые после перерыва производства данного ЛС от трех лет и более;
· в связи с ухудшением их качества.
Процедура предварительного контроля качества ЛС включает следующие этапы:
- направление предприятием – изготовителем в МЗ и СР РФ заявки с необходимыми документами
- анализ документов и выдача разрешения на проведение предварительного КК ЛС
- отбор образцов ЛС для целей предварительного КК
- направление на экспертизу качества образцов ЛС
- принятие МЗ и СР РФ решения по результатам проведенной экспертизы качества ЛС.
Предприятие – изготовитель, впервые начинающее серийный выпуск ЛС, должно отправить на предварительный контроль образцы первых трёх промышленных серий данного препарата.
При изменении наименования ЛС предприятие – изготовитель направляет на предварительный КК одну серию переименованного ЛС.
ЛС снимается с предварительного КК и переводится на выборочный контроль по решению МЗ и СР РФ, если качество всех представленных образцов соответствует требованиям гос. стандарта качества данного ЛС.
2. Выборочный контроль качества.
Выборочному КК подлежат ЛС отечественного и импортного производства, находящие в сфере обращения ЛС в РФ.
Процедура выборочного контроля ЛС включает следующие этапы:
- принятие решения МЗ и СР РФ о проведении выборочного контроля;
- отбор образцов ЛС для целей выборочного контроля;
- направление на экспертизу образцов ЛС;
- принятие решения по результатам экспертизы.
Выборочный контроль качества проводится по показателям «Описание», «Упаковка», «Маркировка», проверяется происхождение, соответствие ЛС сопроводительным документам и государственному стандарту качества.
3. Повторный выборочный контроль качества.
Осуществляется в том случае, если возникают претензии к качеству и безопасности того или иного препарата (серии препаратов) между поставщиком и аптекой (ЛПУ) или аптекой (ЛПУ) и конечным потребителем.
5.3. Порядок проведения сертификации качества лекарственных средств
Законом предусмотрены две формы подтверждения соответствия:
· обязательная - в виде декларирования соответствия и обязательной сертификации,
· добровольная - в виде добровольной сертификации.
Сертификация продукции - это форма подтверждения соответствия продукции требованиям технических регламентов, стандартов или условиям договоров, осуществляемая органом по сертификации, аккредитованном для выполнения данной работы в установленном порядке.
Цели сертификации:
1) создание условий для свободного перемещения товаров по РФ, а также для участия в международной торговле, экономическом и научно-техническом сотрудничестве;
2) повышение конкурентоспособности продукции (работ, услуг) на российском и международном рынках;
3) содействие приобретателю в компетентном выборе продукции (работ, услуг);
4) удостоверение соответствия продукции техническим регламентам, стандартам, условиям договоров.
Сертификация может носить обязательный и добровольный характер.
Обязательная сертификация — производится в случаях, предусмотренных законодательными актами РФ и установленных соответствующим техническим регламентом и исключительно на соответствие требованиям технического регламента.
Подлежат такой процедуре потенциально опасные для приобретателя виды продукции.
Добровольная сертификация - проводится по продукции, не подлежащей обязательной сертификации, по инициативе заявителя на условиях договора между заявителем и органом по сертификации для установления соответствия продукции национальным стандартам, стандартам организаций, системам добровольной сертификации, условиям договоров.
В настоящее время действует постановление Правительства РФ № 1013 от 13.08.1997 (с изм.). Процедура сертификации включает следующие этапы:
1) подача заявки на сертификацию,
2) рассмотрение заявки и принятие решения по ней;
3) проведение проверок (испытаний, проверки производства и т.п.);
4) анализ полученных результатов и принятие решения о выдаче сертификата;
5) выдача сертификата;
6) инспекционный контроль за объектом сертификации органом сертификации с привлечением других организаций.
Сертификат соответствия. Системы сертификации ГОСТ Р (Ростехрегулирования) (СС ГОСТ Р) - это документ, удостоверяющий соответствие продукции (объекта) установленным требованиям (технических регламентов, положениям стандартов, условиям договоров).
В графах СС ГОСТ Р указываются следующие сведения:
1) регистрационный номер СС в Государственном реестре системы сертификации (начинается с букв РОСС, что означает Россия);
2) срок действия сертификата;
3) наименование объекта сертификации (ОС), его регистрационный номер, адрес и телефон;
4) наименование продукции, ссылка на имеющиеся приложение, для партии - размер, номер договора (контракта);
5) коды продукции по общероссийскому классификатору продукции и по классификатору товарной номенклатуры внешней экономической деятельности (для импорта и экспорта);
6) сведения об изготовителе и держателе сертификата (наименование, адрес);
7) номера НД на продукцию;
8) перечень документов, на основании которых выдан СС;
9) дополнительная информация, которую определяет ОС (внешние признаки продукции, условия действия сертификата, место маркировки знаком соответствия, дата инспекционного контроля и т.д.). СС подписывается руководителем ОС и экспертом. Оригинал СС заверяется печатью органа сертификации, копии - установленными способами.
В «Правилах продажи отдельных видов товаров ...», утвержденных Постановлением Правительства РФ № 55 (19.01.98г. в ред. ПП №49 от 01.02.05г.), регламентируются способы заверки копии сертификата. Это может быть один из трех способов:
1) держателем подлинника сертификата;
2) органом по сертификации, выдавшим сертификат;
3) нотариусом.
Кроме того, копии сертификатов, выданных органами сертификации Ростехрегулирования, имеют право заверять территориальные подразделения Ростехрегулирования - Центры стандартизации и метрологии (ЦСМ), а копии сертификатов на ЛС - любые аккредитованные органы сертификации ЛС.
Согласно ПП №55 факт сертификации при розничной реализации
может подтверждаться одним из следующих документов:
1) подлинником сертификата или декларации о соответствии;
2) копией сертификата, заверенной одним из способов;
3) товарно-сопроводительными документами, содержащими по каждому наименованию продукции (а для ЛС - по каждой серии) следующие сведения: номер сертификата соответствия, срок его действия, орган, выдавший сертификат, или регистрационный номер декларации о соответствии, срок ее действия, наименование изготовителя или поставщика (продавца), принявшего декларацию, и орган, ее зарегистрировавший.
Эти документы должны быть заверены подписью и печатью изготовителя (поставщика, продавца) с указанием его адреса и телефона.
Сертификат соответствия на продукцию, которой необходимо проведение проверок другими органами (например, санитарно - эпидемиологической экспертизы), выдается только при наличии соответствующего (например, санитарно - эпидемиологического) заключения, подтверждающего соответствие требованиям безопасности (на марлю, вату, бинты, продукты диетического питания, парфюмерно-косметическую продукцию и средства гигиены полости рта и т.д.). Сведения о наличии такого документа заносятся в бланк сертификата соответствия, поэтому его можно отдельно не предъявлять.
Сертификат действует на всей территории РФ. Срок действия не более 3 лет. На партию или единичное изделие срок действия СС не устанавливается, так как соответствует окончанию срока реализации партии или срока годности изделия.
Для серийно выпускаемой продукции отечественного производства, которая выпущена в период действия СС, сертификат действует до окончания срока годности (службы) продукции данной серии.
Документ о качестве на конкретную серию оформляется производителем, это может быть паспорт ОТК, удостоверение о качестве и безопасности или другой аналогичный документ.
Данные требования распространяются на все виды продукции, подлежащей обязательной сертификации, кроме медицинских иммунобиологических препаратов.
При проведении приемочного контроля следует обращать внимание на:
• срок действия сертификата,
• соответствие сведений о товаре и производителе в СС данным по упаковке и в товарно-сопроводительных документах поставщика,
• на правильность заверки копии сертификата (наличие необходимой оригинальной печати).
При приемке необходимо также помнить, что в соответствии с приказом МЗ РФ №156 от 10.05.2000 применение в медицинских целях изделий медицинского назначения на территории РФ разрешается после их государственной регистрации МЗ РФ или Росздравнадзором. Документом, подтверждающим факт регистрации, является регистрационное удостоверение МЗ РФ или Росдравнадзора, которое необходимо требовать у поставщика.
5.4. Процедура декларирования соответствия лекарственных средств
Переход от жесткой процедуры обязательной сертификации к более доступному и удобному для производителя декларированию соответствия является в последнее время одним из направлений в сфере технического регулирования.
Процедура декларирования соответствия продукции регламентирована ФЗ «О техническом регулировании» и Постановлением Правительства РФ №766 от 07.07.1999 (с изм.) «Об утверждении Перечня продукции, соответствие которой может быть подтверждено декларацией о соответствии, порядка принятия декларации о соответствии и ее регистрации».
Согласно ФЗ № 184 «О техническом регулировании», декларация о соответствии - это документ, удостоверяющий соответствие выпускаемой в обращение продукции требованиям технических регламентов.
Декларирование соответствия осуществляется по одной из следующих схем:
· принятие декларации о соответствии на основании собственных доказательств;
· принятие декларации о соответствии на основании собственных доказательств и доказательств, полученных с участием органа по сертификации и (или) аккредитованной испытательной лаборатории (центра) (далее - третья сторона).
При декларировании соответствия заявителем может быть юридическое лицо или индивидуальный предприниматель, являющиеся изготовителем или продавцом. Круг заявителей устанавливается соответствующим техническим регламентом.
При декларировании соответствия на основании собственных доказательств заявитель использует техническую документацию, результаты собственных исследований (испытаний) и измерений и (или) другие документы.
При декларировании соответствия на основании собственных доказательств и с участием третьей стороны заявитель по своему выбору в дополнение использует протоколы исследований (испытаний) и измерений, проведенных в аккредитованной испытательной лаборатории (центре) или сертификат системы качества.
Декларация о соответствии оформляется на русском языке и должна содержать:
· наименование и местонахождение заявителя;
· наименование и местонахождение изготовителя;
· информацию об объекте подтверждения соответствия, позволяющую идентифицировать этот объект;
· наименование технического регламента, на соответствие требованиям которого подтверждается продукция;
· заявление заявителя о безопасности продукции при ее использовании в соответствии с целевым назначением и принятии заявителем мер по обеспечению соответствия продукции требованиям технических регламентов;
· сведения о проведенных исследованиях (испытаниях) и измерениях, сертификате системы качества, а также документах, послуживших основанием для подтверждения соответствия продукции требованиям технических регламентов;
· срок действия декларации о соответствии (определяется техническим регламентом).
Форма декларации о соответствии утверждается федеральным органом исполнительной власти по техническому регулированию.
Декларация о соответствии принимается на срок, установленный изготовителем (продавцом, исполнителем) продукции исходя из планируемого срока выпуска данной продукции, оказания конкретных услуг или срока.
5.5. Особенности декларирования ЛС
Министерством промышленности и энергетики РФ 26 декабря 2006 г. были утверждены «Методические рекомендации по принятию и регистрации декларации о соответствии лекарственных средств», согласно которым декларированию подлежат лекарственные средства, зарегистрированные в установленном порядке и состоящие из смешанных или несмешанных продуктов для использования в терапевтических целях, расфасованные в виде дозированных лекарственных форм или в упаковке для розничной продажи (коды 931000 -937000 Общероссийского классификатора продукции ОК 005-93).
Декларирование не распространяется на лекарственные средства, изготовленные в аптеках по рецептам врачей, по требованиям учреждений здравоохранения, внутриаптечную заготовку и фасовку, лекарственные средства, предназначенные для проведения клинических исследований (испытаний) или проведения регистрации лекарственных средств в установленном порядке.
В соответствии с действующим законодательством декларации о соответствии на ЛС оформляются с 1 января 2007 г. Лекарственные средства, имеющие сертификат соответствия, выданный в установленном порядке, не подлежат декларированию.
Декларация о соответствии принимается в отношении каждой серии (партии) ЛС на срок, установленный изготовителем (продавцом), но не более установленного срока годности ЛС.
Принятая изготовителем (продавцом) декларация подлежит регистрации в органе по сертификации, аккредитованном в установленном порядке, по выбору изготовителя (продавца).
Регистрация декларации о соответствии осуществляется путем присвоения ей регистрационного номера, содержащего идентификационное обозначение (код) органа по сертификации и порядковый номер декларации по реестру, который ведет орган по сертификации.
ПРИЛОЖЕНИЕ
Нормативные документы
ПРАВИТЕЛЬСТВО РОССИЙСКОЙ ФЕДЕРАЦИИ
ПОСТАНОВЛЕНИЕ
от 1 декабря 2009 г. N 982
ОБ УТВЕРЖДЕНИИ ЕДИНОГО ПЕРЕЧНЯ
ПРОДУКЦИИ, ПОДЛЕЖАЩЕЙ ОБЯЗАТЕЛЬНОЙ СЕРТИФИКАЦИИ,
И ЕДИНОГО ПЕРЕЧНЯ ПРОДУКЦИИ, ПОДТВЕРЖДЕНИЕ СООТВЕТСТВИЯ
КОТОРОЙ ОСУЩЕСТВЛЯЕТСЯ В ФОРМЕ ПРИНЯТИЯ
ДЕКЛАРАЦИИ О СООТВЕТСТВИИ
(извлечение)
(в ред. Постановлений Правительства РФ
от 04.03.2013 N 182,
от 04.10.2013 N 870)
В соответствии с пунктом 3 статьи 46 Федерального закона "О техническом регулировании" Правительство Российской Федерации постановляет:
1. Утвердить прилагаемые:
единый перечень продукции, подлежащей обязательной сертификации;
единый перечень продукции, подтверждение соответствия которой осуществляется в форме принятия декларации о соответствии.
2. Министерству промышленности и торговли Российской Федерации разработать и утвердить в месячный срок порядок представления федеральными органами исполнительной власти информации о продукции, подлежащей обязательному подтверждению соответствия, и ее опубликования.
3. Федеральному агентству по техническому регулированию и метрологии и Федеральной таможенной службе с участием заинтересованных федеральных органов исполнительной власти на основе единых перечней продукции, утвержденных пунктом 1 настоящего Постановления, обеспечить публикацию информации:
о продукции, подлежащей обязательному подтверждению соответствия с указанием нормативных документов, устанавливающих обязательные требования;
о продукции, подлежащей обязательному подтверждению соответствия при помещении под таможенные режимы, предусматривающие возможность отчуждения или использования в соответствии с ее назначением на таможенной территории Российской Федерации, с указанием кодов товарной номенклатуры внешнеэкономической деятельности.
3.1. Сертификаты соответствия на продукцию, выданные до дня вступления в силу настоящего Постановления, считаются действительными до окончания срока, установленного в них, в пределах срока годности или срока службы продукции, установленных в соответствии с законодательством Российской Федерации.
(п. 3.1 введен Постановлением Правительства РФ от 17.03.2010 N 148)
4. Настоящее Постановление не распространяется на отношения, возникающие при проведении оценки соответствия продукции, требования к которой устанавливаются в соответствии со статьей 5 Федерального закона "О техническом регулировании".
5. Признать утратившими силу акты Правительства Российской Федерации по перечню согласно приложению.
6. Настоящее Постановление вступает в силу по истечении 2 месяцев со дня его официального опубликования, за исключением пункта 2, который вступает в силу со дня официального опубликования настоящего Постановления.
Председатель Правительства
Российской Федерации
В.ПУТИН
ПРАВИТЕЛЬСТВО РОССИЙСКОЙ ФЕДЕРАЦИИ
ПОСТАНОВЛЕНИЕ
от 19 января 1998 г. N 55
ОБ УТВЕРЖДЕНИИ
ПРАВИЛ ПРОДАЖИ ОТДЕЛЬНЫХ ВИДОВ
ТОВАРОВ, ПЕРЕЧНЯ ТОВАРОВ ДЛИТЕЛЬНОГО
ПОЛЬЗОВАНИЯ, НА КОТОРЫЕ НЕ РАСПРОСТРАНЯЕТСЯ
ТРЕБОВАНИЕ ПОКУПАТЕЛЯ О БЕЗВОЗМЕЗДНОМ ПРЕДОСТАВЛЕНИИ
ЕМУ НА ПЕРИОД РЕМОНТА ИЛИ ЗАМЕНЫ АНАЛОГИЧНОГО ТОВАРА,
И ПЕРЕЧНЯ НЕПРОДОВОЛЬСТВЕННЫХ ТОВАРОВ НАДЛЕЖАЩЕГО
КАЧЕСТВА, НЕ ПОДЛЕЖАЩИХ ВОЗВРАТУ ИЛИ ОБМЕНУ
НА АНАЛОГИЧНЫЙ ТОВАР ДРУГИХ РАЗМЕРА, ФОРМЫ,
ГАБАРИТА, ФАСОНА, РАСЦВЕТКИ ИЛИ КОМПЛЕКТАЦИИ
(в ред. Постановлений Правительства РФ от 20.10.1998 N 1222,
от 02.10.1999 N 1104, от 06.02.2002 N 81 (ред. 23.05.2006),
от 12.07.2003 N 421, от 01.02.2005 N 49, от 08.02.2006 N 80,
от 15.12.2006 N 770, от 27.03.2007 N 185, от 27.01.2009 N 50,
от 21.08.2012 N 842, от 04.10.2012 N 1007)
(извлечение)
В соответствии с Законом Российской Федерации "О защите прав потребителей" (Собрание законодательства Российской Федерации, 1996, N 3, ст. 140) Правительство Российской Федерации постановляет:
1. Утвердить прилагаемые:
Правила продажи отдельных видов товаров;
Перечень товаров длительного пользования, на которые не распространяется требование покупателя о безвозмездном предоставлении ему на период ремонта или замены аналогичного товара;
Перечень непродовольственных товаров надлежащего качества, не подлежащих возврату или обмену на аналогичный товар других размера, формы, габарита, фасона, расцветки или комплектации.
Председатель Правительства
Российской Федерации
В.ЧЕРНОМЫРДИН
ПРАВИЛА
ПРОДАЖИ ОТДЕЛЬНЫХ ВИДОВ ТОВАРОВ
I. Общие положения
1. Настоящие Правила разработаны в соответствии с Законом Российской Федерации "О защите прав потребителей" и регулируют отношения между покупателями и продавцами при продаже отдельных видов продовольственных и непродовольственных товаров.
2. Под покупателем понимается гражданин, имеющий намерение заказать или приобрести либо заказывающий, приобретающий или использующий товары исключительно для личных, семейных, домашних и иных нужд, не связанных с осуществлением предпринимательской деятельности.
Под продавцом понимается организация независимо от организационно-правовой формы, а также индивидуальный предприниматель, осуществляющие продажу товаров по договору розничной купли-продажи (далее именуется - договор).
3. Режим работы продавца - государственной или муниципальной организации устанавливается по решению соответствующих органов исполнительной власти или органов местного самоуправления.
Режим работы продавца - организации иной организационно-правовой формы, а также индивидуального предпринимателя устанавливается ими самостоятельно.
В случае временного приостановления своей деятельности (для проведения плановых санитарных дней, ремонта и в других случаях) продавец обязан своевременно предоставить покупателю информацию о дате и сроках приостановления деятельности.
4. Ассортимент предлагаемых к продаже товаров, перечень оказываемых услуг, а также формы обслуживания определяются продавцом самостоятельно в соответствии с профилем и специализацией своей деятельности.
При осуществлении розничной торговли в месте нахождения покупателя вне стационарных мест торговли: на дому, по месту работы и учебы, на транспорте, на улице и в иных местах (далее именуется - разносная торговля) не допускается продажа продовольственных товаров (за исключением мороженого, безалкогольных напитков, кондитерских и хлебобулочных изделий в упаковке изготовителя товара), лекарственных препаратов, изделий из драгоценных металлов и драгоценных камней, оружия и патронов к нему, экземпляров аудиовизуальных произведений и фонограмм, программ для электронных вычислительных машин и баз данных.
(абзац введен Постановлением Правительства РФ от 06.02.2002 N 81, в ред. Постановлений Правительства РФ от 12.07.2003 N 421, от 27.03.2007 N 185, от 04.10.2012 N 1007)
5. Продавец при осуществлении своей деятельности обязан соблюдать обязательные требования к организации и осуществлению торговой деятельности, установленные нормативными правовыми актами Российской Федерации.
(п. 5 в ред. Постановления Правительства РФ от 04.10.2012 N 1007)
6. Продавец должен располагать необходимыми помещениями, оборудованием и инвентарем, обеспечивающими в соответствии с законодательством Российской Федерации о техническом регулировании сохранение качества и безопасности товаров при их хранении и реализации в месте продажи, надлежащие условия торговли, а также возможность правильного выбора покупателями товаров.
(в ред. Постановления Правительства РФ от 04.10.2012 N 1007)
7. Продавец обязан иметь и содержать в исправном состоянии средства измерения, своевременно и в установленном порядке проводить их метрологическую поверку.
Для проверки покупателем правильности цены, меры и веса приобретенного товара в торговом зале на доступном месте должно быть установлено соответствующее измерительное оборудование.
8. Продавец обязан иметь книгу отзывов и предложений, которая предоставляется покупателю по его требованию.
9. Настоящие Правила в наглядной и доступной форме доводятся продавцом до сведения покупателей.
10. Продавец обязан довести до сведения покупателя фирменное наименование (наименование) своей организации, место ее нахождения (адрес) и режим работы, размещая указанную информацию на вывеске организации.
(в ред. Постановления Правительства РФ от 04.10.2012 N 1007)
Продавец - индивидуальный предприниматель должен предоставить покупателю информацию о государственной регистрации и наименовании зарегистрировавшего его органа.
Если деятельность, осуществляемая продавцом, подлежит лицензированию, то он обязан предоставить информацию о номере и сроке действия лицензии, а также об органе, ее выдавшем.
Указанная информация размещается в удобных для ознакомления покупателя местах.
Аналогичная информация также должна быть доведена до сведения покупателей при осуществлении торговли во временных помещениях, на ярмарках, с лотков и в других случаях, если торговля осуществляется вне постоянного места нахождения продавца.
При осуществлении разносной торговли представитель продавца должен иметь личную карточку, заверенную подписью лица, ответственного за ее оформление, и печатью продавца, с фотографией, указанием фамилии, имени, отчества представителя продавца, а также сведений о продавце.
(абзац введен Постановлением Правительства РФ от 06.02.2002 N 81)
11. Продавец обязан своевременно в наглядной и доступной форме довести до сведения покупателя необходимую и достоверную информацию о товарах и их изготовителях, обеспечивающую возможность правильного выбора товаров.
Информация в обязательном порядке должна содержать:
наименование товара;
место нахождения (адрес), фирменное наименование (наименование) изготовителя (продавца), место нахождения (адрес) организации (организаций), уполномоченной изготовителем (продавцом) на принятие претензий от покупателей и производящей ремонт и техническое обслуживание товара, для импортного товара - наименование страны происхождения товара;
сведения об обязательном подтверждении соответствия товаров в порядке, определенном законодательством Российской Федерации о техническом регулировании;
сведения об основных потребительских свойствах товара;
сведения об энергетической эффективности товаров, в отношении которых требование о наличии такой информации определено в соответствии с законодательством Российской Федерации об энергосбережении и о повышении энергетической эффективности;
правила и условия эффективного и безопасного использования товара;
гарантийный срок, если он установлен для конкретного товара;
срок службы (срок годности), если он установлен для конкретного товара, а также сведения о необходимых действиях покупателя по истечении указанного срока и возможных последствиях при невыполнении таких действий, если товары по истечении указанного срока представляют опасность для жизни, здоровья и имущества покупателя или становятся непригодными для использования по назначению;
цену в рублях и условия приобретения товаров, в том числе при предоставлении кредита - размер кредита, полную сумму, подлежащую выплате потребителем, и график погашения этой суммы.
Если приобретаемый покупателем товар был в употреблении или в нем устранялся недостаток (недостатки), покупателю должна быть предоставлена информация об этом.
Об имеющихся в товаре недостатках продавец должен предупредить покупателя не только в устной, но и в письменной форме (на ярлыке товара, товарном чеке или иным способом).
(п. 11 в ред. Постановления Правительства РФ от 04.10.2012 N 1007)
12. Продавец обязан по требованию потребителя ознакомить его с товарно-сопроводительной документацией на товар, содержащей по каждому наименованию товара сведения об обязательном подтверждении соответствия согласно законодательству Российской Федерации о техническом регулировании (сертификат соответствия, его номер, срок его действия, орган, выдавший сертификат, или сведения о декларации о соответствии, в том числе ее регистрационный номер, срок ее действия, наименование лица, принявшего декларацию, и орган, ее зарегистрировавший). Эти документы должны быть заверены подписью и печатью поставщика или продавца с указанием его места нахождения (адреса) и телефона.
(п. 12 в ред. Постановления Правительства РФ от 04.10.2012 N 1007)
13. Продажа товаров, изготовленных из объектов животного мира (меховые и кожаные швейные, галантерейные, декоративные изделия, обувь, пищевые продукты), принадлежащих к видам, занесенным в Красную книгу Российской Федерации, осуществляется при наличии соответствующей документации на товары, подтверждающей, что эти объекты животного мира добыты в соответствии с законодательством Российской Федерации на основании разрешения (распорядительной лицензии), выдаваемого федеральным органом исполнительной власти в области охраны окружающей природной среды. Продажа ввезенных в Российскую Федерацию товаров, изготовленных из объектов животного мира, подпадающих под действие Конвенции о международной торговле видами дикой фауны и флоры, находящимися под угрозой исчезновения, осуществляется на основании разрешения компетентного органа страны экспортера, а товаров, конфискованных в результате нарушения указанной Конвенции, - на основании разрешения уполномоченного органа.
(в ред. Постановления Правительства РФ от 06.02.2002 N 81)
При продаже таких товаров продавец обязан предоставить покупателю по его просьбе сведения о документах, подтверждающих наличие соответствующего разрешения.
14. Продавец должен предоставить также другую информацию о товарах, предусмотренную федеральными законами, иными нормативными правовыми актами Российской Федерации.
(в ред. Постановления Правительства РФ от 04.10.2012 N 1007)
15. Информация о товаре, его изготовителе и продавце должна доводиться до сведения покупателя способами, установленными федеральными законами, иными нормативными правовыми актами Российской Федерации, а если указанными актами они не определены, то способами, принятыми для отдельных видов товаров.
(в ред. Постановления Правительства РФ от 04.10.2012 N 1007)
Объем обязательной информации о товаре, его изготовителе, передаваемой покупателю вместе с товаром (на товаре, потребительской таре, упаковке, ярлыке, этикетке, в технической документации), должен соответствовать требованиям федеральных законов, иных нормативных правовых актов Российской Федерации.
(в ред. Постановления Правительства РФ от 04.10.2012 N 1007)
Информация о продавце, товарах и их изготовителях доводится до сведения покупателей на русском языке, а дополнительно, по усмотрению продавца, на государственных языках субъектов Российской Федерации и языках народов Российской Федерации.
16. Потребителю также должна быть предоставлена наглядная и достоверная информация об оказываемых услугах, ценах на них и условиях оказания услуг, а также о применяемых формах обслуживания при продаже товаров (по предварительным заказам, продажа товаров на дому и другие формы).
17. При продаже товаров покупателю предоставляется возможность самостоятельно или с помощью продавца ознакомиться с необходимыми товарами.
Покупатель вправе осмотреть предлагаемый товар, потребовать проведения в его присутствии проверки свойств или демонстрации его действия, если это не исключено ввиду характера товара и не противоречит правилам, принятым в розничной торговле.
Продавец обязан проводить проверку качества и безопасности (осмотр, испытание, анализ, экспертизу) предлагаемого для продажи товара в случае, когда проведение проверок предусмотрено законодательством Российской Федерации или условиями договора.
(в ред. Постановления Правительства РФ от 04.10.2012 N 1007)
18. Цены товаров, реализуемых продавцом, а также иные условия договора должны быть одинаковыми для всех покупателей, за исключением случаев, когда федеральными законами или иными нормативными правовыми актами допускается предоставление льгот для отдельных категорий покупателей.
19. Продавец обязан обеспечить наличие единообразных и четко оформленных ценников на реализуемые товары с указанием наименования товара, сорта (при его наличии), цены за вес или единицу товара, подписи материально ответственного лица или печати организации, даты оформления ценника.
(в ред. Постановления Правительства РФ от 04.10.2012 N 1007)
При продаже товаров, осуществляемой посредством разносной торговли, представитель продавца обязан иметь прейскурант, заверенный подписью лица, ответственного за его оформление, и печатью продавца, с указанием наименования и цены товаров, а также предоставляемых с согласия покупателя услуг.
(абзац введен Постановлением Правительства РФ от 06.02.2002 N 81)
20. Договор считается заключенным в надлежащей форме с момента выдачи продавцом покупателю кассового или товарного чека или иного документа, подтверждающего оплату товара, если иное не предусмотрено федеральным законом или договором между продавцом и покупателем.
При разносной торговле вместе с товаром (за исключением продовольственных товаров, указанных в абзаце втором пункта 4 настоящих Правил) покупателю передается товарный чек, в котором указываются наименование товара и сведения о продавце, дата продажи, количество и цена товара, а также проставляется подпись представителя продавца.
(абзац введен Постановлением Правительства РФ от 06.02.2002 N 81)
21. Расчеты с покупателями за товары осуществляются с применением контрольно-кассовых машин, за исключением предусмотренных законодательством Российской Федерации случаев.
22. Предлагаемые продавцом услуги в связи с продажей товаров могут оказываться только с согласия покупателя.
Покупатель вправе отказаться от услуг, предлагаемых при продаже товара, а также потребовать от продавца возврата сумм, уплаченных за услуги, предоставленные без его согласия.
Продавец не вправе обуславливать продажу одних товаров обязательным приобретением других товаров или обязательным оказанием услуг в связи с их продажей, за исключением случаев, когда товары по техническим требованиям не могут быть собраны и (или) установлены (подключены) без участия соответствующих специалистов.
В случае доставки крупногабаритного товара силами покупателя продавец обязан бесплатно обеспечить погрузку товара на транспортное средство покупателя.
23. Продавец обязан передать покупателю товар надлежащего качества, в таре и (или) упаковке за исключением товара, который по своему характеру не требует затаривания и (или) упаковки, в определенном наборе (комплект товаров) и комплектности, с относящимися к товару документами и принадлежностями.
(в ред. Постановления Правительства РФ от 06.02.2002 N 81)
Требования к качеству, таре и (или) упаковке передаваемого товара, его комплектности, принадлежностям и документации, комплекту товаров, а также к условиям доставки товара устанавливаются законодательством Российской Федерации.
24. Товар, на который установлен срок годности, продавец обязан передать покупателю с таким расчетом, чтобы он мог быть использован по назначению до истечения срока годности.
25. При продаже товара с условием о его принятии покупателем в определенный срок продавец не может продать товар другому покупателю в течение этого срока.
Если иное не предусмотрено договором между продавцом и покупателем, неявка покупателя или несовершение иных необходимых действий для принятия товара в течение определенного договором срока могут рассматриваться продавцом в качестве отказа покупателя от приобретения товара.
26. Покупатель вправе в течение 14 дней с момента передачи ему непродовольственного товара надлежащего качества, если более длительный срок не объявлен продавцом, обменять в месте покупки и иных местах, объявленных продавцом, купленный товар на аналогичный товар других размера, формы, габарита, фасона, расцветки или комплектации, произведя в случае разницы в цене необходимый перерасчет с продавцом.
При отсутствии у продавца необходимого для обмена товара покупатель вправе возвратить приобретенный товар продавцу и получить уплаченную за него денежную сумму или обменять его на аналогичный товар при первом поступлении соответствующего товара в продажу. Продавец обязан сообщить покупателю, потребовавшему обмена непродовольственного товара, о его поступлении в продажу.
Требование покупателя об обмене либо возврате товара подлежит удовлетворению, если товар не был в употреблении, сохранены его товарный вид, потребительские свойства, пломбы, ярлыки, а также имеются доказательства приобретения товара у данного продавца, за исключением товаров, не подлежащих обмену или возврату по указанным в настоящем пункте основаниям в соответствии с перечнем, утверждаемым Правительством Российской Федерации.
27. Покупатель, которому продан товар ненадлежащего качества, если его недостатки не были оговорены продавцом, вправе по своему выбору потребовать от продавца:
замены на товар аналогичной марки (модели, артикула);
замены на такой же товар другой марки (модели, артикула) с соответствующим перерасчетом покупной цены;
соразмерного уменьшения покупной цены;
незамедлительного безвозмездного устранения недостатков товара;
возмещения расходов, понесенных покупателем или третьим лицом, на устранение недостатков товара.
(в ред. Постановления Правительства РФ от 06.02.2002 N 81)
При этом покупатель вправе потребовать также полного возмещения убытков, причиненных ему вследствие продажи товара ненадлежащего качества.
Покупатель вправе требовать замены технически сложного или дорогостоящего товара в случае существенного нарушения требований к его качеству (обнаружения неустранимых недостатков, недостатков, которые не могут быть устранены без несоразмерных расходов или затрат времени, либо выявляются неоднократно, либо проявляются вновь после их устранения, и других подобных недостатков).
В отношении технически сложных товаров указанное требование покупателя подлежит удовлетворению согласно Перечню таких товаров, утверждаемому Правительством Российской Федерации.
В случае обнаружения недостатков товара, свойства которого не позволяют устранить их (продовольственные товары, парфюмерно-косметические изделия, товары бытовой химии и другие товары), покупатель вправе по своему выбору потребовать замены такого товара товаром надлежащего качества либо соразмерного уменьшения покупной цены.
Вместо предъявления указанных требований покупатель вправе отказаться от приобретенного товара и потребовать возврата уплаченной за товар денежной суммы.
При этом покупатель по требованию продавца и за его счет должен возвратить полученный товар ненадлежащего качества.
При возврате покупателю уплаченной за товар денежной суммы продавец не вправе удерживать из нее сумму, на которую понизилась стоимость товара из-за его полного или частичного использования, потери им товарного вида или других подобных обстоятельств.
В случае предъявления покупателем требования об устранении продавцом недостатков товара длительного пользования или замены такого товара покупатель вправе одновременно потребовать предоставление ему на период ремонта или замены товара ненадлежащего качества аналогичного товара надлежащего качества, за исключением товаров по Перечню, утверждаемому Правительством Российской Федерации, на которые это требование не распространяется.
28. Продавец или организация, выполняющая функции продавца на основании договора с ним, обязаны принять товар ненадлежащего качества у покупателя, а в случае необходимости провести проверку качества товара. Покупатель вправе участвовать в проверке качества товара.
При возникновении спора о причинах появления недостатков товара продавец или организация, выполняющая функции продавца на основании договора с ним, обязаны провести экспертизу товара за свой счет. Покупатель вправе оспорить заключение такой экспертизы в судебном порядке.
(в ред. Постановления Правительства РФ от 06.02.2002 N 81)
Отсутствие у покупателя кассового или товарного чека либо иного документа, удостоверяющего факт и условия покупки товара, не является основанием для отказа в удовлетворении его требований и не лишает его возможности ссылаться на свидетельские показания в подтверждение заключения договора и его условий.
(абзац введен Постановлением Правительства РФ от 06.02.2002 N 81)
29. Сроки удовлетворения продавцом требований покупателя, а также ответственность за нарушение этих сроков определяются в соответствии с Законом Российской Федерации "О защите прав потребителей".
30. Покупатель вправе предъявить указанные в пункте 27 настоящих Правил требования в отношении недостатков товара, если они обнаружены в течение гарантийного срока или срока годности.
Гарантийный срок товара, а также срок его службы исчисляется со дня продажи товара покупателю. Если день продажи товара установить невозможно, этот срок исчисляется со дня изготовления товара.
(в ред. Постановления Правительства РФ от 06.02.2002 N 81)
Срок годности товара определяется периодом, исчисляемым со дня изготовления товара, в течение которого он пригоден к использованию, или датой, до наступления которой товар пригоден к использованию.
Если покупатель лишен возможности использовать товар вследствие обстоятельств, зависящих от продавца (товар нуждается в специальной установке, подключении или сборке, в нем имеются недостатки и др.), гарантийный срок исчисляется с даты устранения продавцом таких обстоятельств. Если день доставки, установки, подключения, сборки товара установить невозможно, гарантийный срок исчисляется со дня заключения договора купли-продажи.
(в ред. Постановления Правительства РФ от 06.02.2002 N 81)
Для сезонных товаров (одежда, меховые товары, обувь и другие товары) гарантийный срок исчисляется с момента наступления соответствующего сезона, срок наступления которого определяется уполномоченным государственным органом субъекта Российской Федерации, исходя из климатических условий места нахождения покупателей.
В случае если гарантийный срок составляет менее двух лет и недостатки товара обнаружены покупателем по истечении гарантийного срока, но в пределах двух лет, продавец несет ответственность, если покупатель докажет, что недостатки товара возникли до его передачи покупателю или по причинам, возникшим до этого момента.
(в ред. Постановления Правительства РФ от 06.02.2002 N 81)
31. Если на товар не установлен гарантийный срок или срок годности, требования, связанные с недостатками товара, могут быть предъявлены покупателем при условии, что недостатки обнаружены в разумный срок, но в пределах двух лет со дня передачи товара покупателю либо в пределах более длительного срока, установленного в соответствии с федеральным законом или договором.
(в ред. Постановления Правительства РФ от 06.02.2002 N 81)
VIII. Особенности продажи лекарственных препаратов
и изделий медицинского назначения
(введен Постановлением Правительства РФ
от 20.10.1998 N 1222)
70. Продажа лекарственных препаратов (дозированных лекарственных средств, готовых к применению и предназначенных для профилактики, диагностики и лечения заболеваний человека и животных, предотвращения беременности, повышения продуктивности животных) осуществляется в соответствии с Федеральным законом "Об обращении лекарственных средств" и с учетом особенностей, определенных настоящими Правилами.
(в ред. Постановления Правительства РФ от 04.10.2012 N 1007)
71. Информация о лекарственных препаратах помимо сведений, указанных в пунктах 11 и 12 настоящих Правил, а также предусмотренных статьей 46 Федерального закона "Об обращении лекарственных средств", должна содержать сведения о государственной регистрации лекарственного препарата с указанием номера и даты его государственной регистрации (за исключением лекарственных препаратов, изготовленных продавцом (аптечным учреждением) по рецептам врачей).
(в ред. Постановления Правительства РФ от 04.10.2012 N 1007)
72. Информация об изделиях медицинского назначения (изделиях медицинской техники, включая инструменты, оборудование, приборы и аппараты медицинские, изделия медицинские из резины, текстиля, стекла, полимерных и других материалов, и запасных частях к ним, предназначенных для профилактики, диагностики, лечения заболеваний в домашних условиях, реабилитации и ухода за больными; оправах для корригирующих очков и линзах для коррекции зрения; изделиях протезно-ортопедических и запасных частях к ним; наборах реагентов и средств для диагностики; домашних (автомобильных) аптечных комплектах (наборах) и прочих медицинских материалах и средствах) помимо сведений, указанных в пунктах 11 и 12 настоящих Правил, должна содержать сведения о номере и дате разрешения на применение таких изделий в медицинских целях, выданного Федеральной службой по надзору в сфере здравоохранения в установленном порядке, а также, с учетом особенностей конкретного вида товара, сведения о его назначении, способе и условиях применения, действии и оказываемом эффекте, ограничениях (противопоказаниях) для применения.
(в ред. Постановлений Правительства РФ от 01.02.2005 N 49, от 04.10.2012 N 1007)
73. Продавец должен предоставить покупателю информацию о правилах отпуска лекарственных препаратов и изделий медицинского назначения.
74. Продавец обязан обеспечить продажу лекарственных препаратов минимального ассортимента, необходимых для оказания медицинской помощи, перечень которых устанавливается Министерством здравоохранения Российской Федерации.
(в ред. Постановлений Правительства РФ от 01.02.2005 N 49, от 04.10.2012 N 1007)
75. Лекарственные препараты и изделия медицинского назначения до подачи в торговый зал должны пройти предпродажную подготовку, которая включает распаковку, рассортировку и осмотр товара; проверку качества товара (по внешним признакам) и наличия необходимой информации о товаре и его изготовителе (поставщике).
Предпродажная подготовка изделий медицинской техники включает при необходимости также удаление заводской смазки, проверку комплектности, сборку и наладку.
76. Продажа лекарственных препаратов производится на основании предъявляемых покупателями рецептов врачей, оформленных в установленном порядке, а также без рецептов в соответствии с инструкцией по применению лекарственных препаратов.
(в ред. Постановлений Правительства РФ от 01.02.2005 N 49, от 04.10.2012 N 1007).
МИНИСТЕРСТВО ЗДРАВООХРАНЕНИЯ И СОЦИАЛЬНОГО РАЗВИТИЯ
РОССИЙСКОЙ ФЕДЕРАЦИИ
ПРИКАЗ
от 30 октября 2006 г. N 734
ОБ УТВЕРЖДЕНИИ АДМИНИСТРАТИВНОГО РЕГЛАМЕНТА
ФЕДЕРАЛЬНОЙ СЛУЖБЫ ПО НАДЗОРУ В СФЕРЕ ЗДРАВООХРАНЕНИЯ
И СОЦИАЛЬНОГО РАЗВИТИЯ ПО ИСПОЛНЕНИЮ ГОСУДАРСТВЕННОЙ
ФУНКЦИИ ПО ОРГАНИЗАЦИИ ПРОВЕДЕНИЯ ЭКСПЕРТИЗЫ
КАЧЕСТВА, ЭФФЕКТИВНОСТИ И БЕЗОПАСНОСТИ
ЛЕКАРСТВЕННЫХ СРЕДСТВ
(извлечение)
В соответствии с Постановлением Правительства Российской Федерации от 11 ноября 2005 г. N 679 "О порядке разработки и утверждения административных регламентов исполнения государственных функций и административных регламентов предоставления государственных услуг" (Собрание законодательства Российской Федерации, 2005, N 47, ст. 4933) и Положением о Министерстве здравоохранения и социального развития Российской Федерации, утвержденным Постановлением Правительства Российской Федерации от 30 июня 2004 г. N 321 "Об утверждении Положения о Министерстве здравоохранения и социального развития Российской Федерации" (Собрание законодательства Российской Федерации, 2004, N 28, ст. 2898; 2005, N 2, ст. 162; 2006, N 19, ст. 2080), приказываю:
1. Утвердить прилагаемый Административный регламент Федеральной службы по надзору в сфере здравоохранения и социального развития по исполнению государственной функции по организации проведения экспертизы качества, эффективности и безопасности лекарственных средств.
2. Федеральной службе по надзору в сфере здравоохранения и социального развития (Р.У. Хабриев) обеспечить проведение экспертизы качества, эффективности и безопасности лекарственных средств в соответствии с Административным регламентом, утвержденным настоящим Приказом.
3. Признать утратившими силу Приказы Министерства здравоохранения Российской Федерации от 4 апреля 2003 г. N 137 "Об утверждении Порядка осуществления государственного контроля качества лекарственных средств на территории Российской Федерации" (зарегистрирован Минюстом России 10 апреля 2003 г. N 4399) и от 28 мая 2003 г. N 223 "Об утверждении Положения о порядке проведения государственного контроля эффективности и безопасности лекарственных средств на территории Российской Федерации" (зарегистрирован Минюстом России 3 июня 2003 г. N 4633).
4. Контроль за исполнением настоящего Приказа возложить на заместителя Министра здравоохранения и социального развития Российской Федерации В.И. Стародубова. Министр М.Ю.ЗУРАБОВ
АДМИНИСТРАТИВНЫЙ РЕГЛАМЕНТ
ФЕДЕРАЛЬНОЙ СЛУЖБЫ ПО НАДЗОРУ В СФЕРЕ
ЗДРАВООХРАНЕНИЯ И СОЦИАЛЬНОГО РАЗВИТИЯ ПО ИСПОЛНЕНИЮ
ГОСУДАРСТВЕННОЙ ФУНКЦИИ ПО ОРГАНИЗАЦИИ ПРОВЕДЕНИЯ
ЭКСПЕРТИЗЫ КАЧЕСТВА, ЭФФЕКТИВНОСТИ И БЕЗОПАСНОСТИ
ЛЕКАРСТВЕННЫХ СРЕДСТВ
I. Общие положения
1.1. Административный регламент Федеральной службы по надзору в сфере здравоохранения и социального развития по исполнению государственной функции по организации проведения экспертизы качества, эффективности и безопасности лекарственных средств (далее - Регламент) разработан с учетом требований Федерального закона от 22.06.98 N 86-ФЗ "О лекарственных средствах" (Собрание законодательства Российской Федерации, 1998, N 26, ст. 3006), в соответствии с Постановлением Правительства Российской Федерации от 11.11.2005 N 679 "О порядке разработки и утверждения административных регламентов исполнения государственных функций и административных регламентов предоставления государственных услуг" (Собрание законодательства Российской Федерации, 21.11.2005, N 47, ст. 4933) и в рамках полномочий, установленных Положением о Федеральной службе по надзору в сфере здравоохранения и социального развития, утвержденным Постановлением Правительства Российской Федерации от 30.06.2004 N 323 (Собрание законодательства Российской Федерации, 2004, N 28, ст. 2900).
1.2. Организация проведения экспертизы качества, эффективности и безопасности лекарственных средств представляет собой функцию Федеральной службы по надзору в сфере здравоохранения и социального развития по привлечению научных, иных организаций, ученых и специалистов, для проработки вопросов качества, эффективности и безопасности лекарственных средств посредством проведения исследований, анализа и оценки объектов экспертизы, подготовки заключений относительно этих объектов.
При возникновении необходимости в экспертизе качества, эффективности и безопасности лекарственных средств в ходе проведения мероприятий по государственному контролю в сфере обращения лекарственных средств ее организацию осуществляет Федеральная служба по надзору в сфере здравоохранения и социального развития в соответствии с требованиями законодательства Российской Федерации. При организации проведения экспертизы качества, эффективности и безопасности лекарственных средств не допускается передача каких-либо государственных функций экспертным организациям или экспертам.
1.3. Основными принципами организации экспертизы качества, эффективности и безопасности лекарственных средств являются:
1) независимость и правовая защищенность субъектов экспертизы при осуществлении ими своей профессиональной деятельности;
2) научный подход, полнота, всесторонность и объективность исследований объектов экспертизы, обеспечение обоснованности результатов экспертизы в соответствии с документально установленными критериями приемлемости;
3) компетентность и высокий профессиональный уровень экспертных организаций и экспертов;
4) системность организации экспертной работы и ее методического обеспечения;
5) ориентация на мировой уровень развития науки и техники, нормы и правила экологической, технической и общественной безопасности, на обязательность выполнения требований законодательства Российской Федерации и применимых международных и национальных стандартов;
6) гласность результатов экспертизы при условии сохранения государственной, служебной и коммерческой тайны в соответствии с законодательством Российской Федерации.
1.4. Все документы при проведении экспертизы качества, эффективности и безопасности лекарственных средств должны исполняться на русском языке.
1.5. Научные и иные организации, ученые и специалисты (далее - экспертные организации и эксперты, соответственно) привлекаются Федеральной службой по надзору в сфере здравоохранения и социального развития для проведения экспертизы качества, эффективности и безопасности лекарственных средств на основе договоров.
Проводящие экспертизу качества, эффективности и безопасности лекарственных средств экспертные организации (эксперты) не должны иметь заинтересованность в результатах экспертизы.
1.6. При организации экспертизы, в целях обеспечения ее результативности, качества и соблюдения основных принципов проведения, Федеральная служба по надзору в сфере здравоохранения и социального развития принимает во внимание компетентность и профессионализм экспертных организаций и экспертов, системность организации экспертной работы, обеспечивает методическое сопровождение деятельности по проведению экспертизы качества, эффективности и безопасности лекарственных средств в порядке, определяемом условиями заключенных договоров и внутренними распорядительными актами Федеральной службы по надзору в сфере здравоохранения и социального развития.
1.7. Экспертиза качества, эффективности и безопасности лекарственных средств проводится на основе заданий на проведение экспертизы, определяющих объект (объекты) экспертизы и критерии его (их) оценки. Задания на проведение экспертизы могут быть общими и частными. Общее задание выдается экспертной организации на определенный период времени и устанавливает ее полномочия по проведению определенного вида экспертизы в течение указанного срока. Частное задание выдается экспертной организации на разовое проведение конкретного вида экспертизы. Задания на проведение экспертизы утверждаются руководителем Федеральной службы по надзору в сфере здравоохранения и социального развития или должностным лицом, специально уполномоченным для этой цели.
1.8. Частное задание на проведение экспертизы качества, эффективности и безопасности лекарственных средств должно включать в себя:
1) определение вида и объекта экспертизы;
2) срок проведения экспертизы;
3) возможные особенности проведения экспертизы;
4) другие сведения, необходимые для проведения экспертизы.
1.9. Общее задание на проведение экспертизы качества, эффективности и безопасности лекарственных средств должно включать в себя:
1) определение вида и объекта экспертизы;
2) срок действия полномочий экспертной организации (или эксперта);
3) правила проведения данного вида экспертных работ;
4) другие сведения, необходимые для проведения экспертизы.
1.10. Заключением экспертизы качества, эффективности и безопасности лекарственных средств является документ, содержащий основные выводы по объекту экспертизы. Заключение должно быть обоснованным и недвусмысленно отвечать на поставленные в задании перед экспертной организацией (экспертом) вопросы. К заключению должны (при наличии) прилагаться изложенные в произвольной форме особые мнения экспертов, не согласных с принятым заключением.
Заключение экспертизы по объекту ее проведения может быть положительным или отрицательным, о чем в постановляющей части заключения должна быть соответствующая формулировка.
Заключение экспертизы должно храниться в течение срока не менее 10 лет при условии, что большие сроки не устанавливаются иными нормативными правовыми актами.
По решению руководителя Федеральной службы по надзору в сфере здравоохранения и социального развития информация, содержащаяся в заключении экспертизы качества, эффективности и безопасности лекарственных средств, может публиковаться при условии соблюдения требований по безопасности сведений, составляющих государственную, служебную, коммерческую или иную тайну.
1.11. Серьезными нарушениями правил проведения экспертизы качества, эффективности и безопасности лекарственных средств являются:
1) непредставление экспертной организации (эксперту) информации, необходимой для проведения экспертизы;
2) фальсификация материалов, сведений и данных, представляемых на экспертизу, а также сведений о результатах ее проведения;
3) принуждение экспертной организации (эксперта) к подготовке заведомо ложного заключения экспертизы;
4) создание препятствий проведению экспертизы;
5) необоснованность выводов экспертизы;
6) фальсификация выводов экспертизы;
7) сокрытие от Федеральной службы по надзору в сфере здравоохранения и социального развития оснований для отвода экспертной организации (эксперта) вследствие возникновения заинтересованности экспертной организации (эксперта) в результатах экспертизы;
8) прямое или косвенное вмешательство в процесс экспертизы в целях оказания влияния на ход и результаты экспертизы;
9) иные нарушения, ответственность за которые наступает в соответствии с законодательством Российской Федерации.
1.12. В целях обеспечения возможности организации проведения экспертизы качества в виде выборочного государственного контроля лекарственных средств организации - производители и импортеры должны ежемесячно направлять в Федеральную службу по надзору в сфере здравоохранения и социального развития перечень серий лекарственных средств, выпущенных ими в обращение на территории Российской Федерации.
1.13. Организация проведения экспертизы качества, эффективности и безопасности лекарственных средств включает в себя следующие административные процедуры:
1) организация проведения экспертизы качества, эффективности и безопасности, осуществляемой в ходе рассмотрения документов и принятия решения о государственной регистрации лекарственного средства (при регистрации лекарственных средств). Основание: статья 19 Федерального закона от 22.06.98 N 86-ФЗ "О лекарственных средствах"; п. 5.2 Положения о Федеральной службе по надзору в сфере здравоохранения и социального развития", утвержденного Постановлением Правительства Российской Федерации от 30.06.2004 N 323;
2) осуществление сбора и анализа информации о побочных эффектах применения лекарственных средств. Основания: статья 41 Федерального закона от 22.06.98 N 86-ФЗ "О лекарственных средствах"; п. 5.1.3.5 "Положения о Федеральной службе по надзору в сфере здравоохранения и социального развития", утвержденного Постановлением Правительства Российской Федерации от 30.06.2004 N 323;
3) организация проведения экспертизы качества при осуществлении предварительного государственного контроля лекарственных средств. Основание: статья 8 Федерального закона от 22.06.98 N 86-ФЗ "О лекарственных средствах". Осуществляется для впервые производимых и впервые ввозимых на территорию Российской Федерации лекарственных средств; для лекарственных средств, выпускаемых по измененной технологии; для лекарственных средств, выпускаемых после перерыва производства данного лекарственного средства от трех лет и более; для иных лекарственных средств вследствие выявления ухудшения их качества;
4) организация проведения экспертизы качества при осуществлении выборочного государственного контроля лекарственных средств. Основание: статья 8 Федерального закона от 22.06.98 N 86-ФЗ "О лекарственных средствах". Осуществляется на основании планов выборочного контроля качества Федеральной службы по надзору в сфере здравоохранения и социального развития, формируемого по результатам сбора и анализа информации о качестве лекарственных средств;
5) организация проведения экспертизы качества при осуществлении повторного выборочного государственного контроля лекарственных средств. Основание: статья 8 Федерального закона от 22.06.98 N 86-ФЗ "О лекарственных средствах". Осуществляется в случае возникновения сомнений в качестве лекарственных средств у субъекта обращения лекарственных средств;
6) осуществление сбора и анализа информации о качестве лекарственных средств. Основание: п. 5.1.3.5 "Положения о Федеральной службе по надзору в сфере здравоохранения и социального развития", утвержденного Постановлением Правительства Российской Федерации от 30.06.2004 N 323.
II. Требования к порядку исполнения государственной функции.
2.1. Порядок информирования о государственной функции по организации проведения экспертизы качества, эффективности и безопасности лекарственных средств:
2.1.1. Документом, результирующим проведение экспертизы качества, эффективности и безопасности лекарственного средства, является экспертное заключение, которое используется Федеральной службой по надзору в сфере здравоохранения и социального развития при исполнении соответствующих государственных контрольно-надзорных функций.
2.1.2. Направление документов и данных для организации проведения экспертизы качества, эффективности и безопасности лекарственных средств производится по адресу:
Федеральная служба по надзору в сфере здравоохранения и социального развития, отдел, осуществляющий регистрацию лекарственных средств: 109074, Москва, Славянская площадь, д. 4, строение 1. Время работы: в будние дни с 9-00 до 18-00.
Место приема документов должно быть оснащено телефоном, компьютером с возможностью выхода в Интернет и текстом настоящего регламента.
Телефоны для справок и предварительной записи: +7(495) 298-4070; +7(495) 298-4069.
Адрес электронной почты: общий: qc@roszdravnadzor.ru; для передачи организациями перечней серий лекарственных средств, выпущенных ими в обращение на территории Российской Федерации: qc-release@roszdravnadzor.ru.
Общая справочная служба: +7(495) 298-4628.
Информация об итогах проведения экспертизы качества, эффективности и безопасности лекарственных средств может быть получена на официальном Интернет-сайте: www.roszdravnadzor.ru.
2.1.3. Перечни документов, представляемых для организации проведения экспертизы качества, эффективности и безопасности лекарственных средств, и требования к таким документам представлены в соответствующих разделах административных процедур настоящего Регламента.
2.2. Условия и сроки исполнения государственной функции по организации проведения экспертизы качества, эффективности и безопасности лекарственных средств представлены в соответствующих разделах административных процедур настоящего Регламента.
Организация проведения экспертизы качества лекарственного средства в виде предварительного государственного контроля осуществляется в срок, не превышающий 40 дней с даты отбора образцов, но не более 50 дней с даты получения комплекта соответствующих документов и данных (без учета времени, затраченного обратившейся организацией на наработку трех опытно-промышленных серий лекарственного средства) при условии соблюдения требований к их составу и содержанию.
Организация проведения экспертизы качества лекарственного средства в виде повторного выборочного государственного контроля осуществляется в срок, не превышающий 40 дней с даты отбора образцов, но не более 50 дней с даты получения комплекта соответствующих документов и данных при условии соблюдения требований к их составу и содержанию.
2.3. Основания для отказа в рассмотрении документов или организации проведения экспертизы качества, эффективности и безопасности лекарственных средств содержатся в соответствующих разделах административных процедур настоящего Регламента.
2.4. Действия или бездействие Федеральной службы по надзору в сфере здравоохранения и социального развития в связи с организацией проведения экспертизы качества, эффективности и безопасности лекарственных средств могут быть обжалованы в установленном порядке. Министр здравоохранения и социального развития Российской Федерации отменяет противоречащие федеральному законодательству решения Федеральной службы по надзору в сфере здравоохранения и социального развития, если иной порядок отмены решений не установлен федеральным законом.
III. Административные процедуры
3.1. Структура и взаимосвязи административных процедур, выполняемых при организации проведения экспертизы качества, эффективности и безопасности лекарственных средств, приведены на схеме (Приложение 1).
3.2. Руководители подразделений Федеральной службы по надзору в сфере здравоохранения и социального развития, отвечающих в соответствии с настоящим Регламентом за организацию проведения экспертизы качества, эффективности и безопасности лекарственных средств, должны организовать документированный учет выполнения каждого этапа административных процедур с указанием даты завершения его исполнения и подписи ответственного лица.
3.3. Административная процедура "Организация проведения экспертизы качества, эффективности и безопасности лекарственных средств (при государственной регистрации)" осуществляется по необходимости только в ходе выполнения административных процедур "Рассмотрение документов и принятие решения о государственной регистрации лекарственных средств", "Внесение изменений в регистрационную документацию на лекарственные средства", "Рассмотрение фактов и обстоятельств, создающих угрозу для жизни и здоровья людей при применении зарегистрированных лекарственных средств" административного регламента Федеральной службы по надзору в сфере здравоохранения и социального развития "Государственная регистрация лекарственных средств" в соответствии с нижеследующим порядком (схема осуществления административной процедуры приведена в Приложении 2):
3.3.1. Экспертиза качества и (или) эффективности и (или) безопасности лекарственных средств производится при наличии следующих оснований:
- при отсутствии достаточных экспертных заключений в отношении данных о производстве лекарственного средства и (или) методов контроля качества лекарственного средства и (или) результатов доклинических исследований лекарственного средства и (или) результатов фармакологических и токсикологических исследований лекарственного средства и (или) результатов клинических исследований лекарственного средства (с учетом целей проведения экспертизы);
- при недостаточной обоснованности и (или) неоднозначном характере представленных в документах и данных результатов оценки качества, эффективности и безопасности лекарственного средства;
- при противоречивости имеющихся экспертных заключений (производится обязательное назначение дополнительной экспертизы).
3.3.2. При наличии оснований должностное лицо, отвечающее за выполнение соответствующей процедуры (ответственный исполнитель), готовит проект задания на проведение экспертизы качества, эффективности и безопасности лекарственного средства с указанием организации(й), которая(ые) будет(ут) проводить экспертизу, предельных сроков выполнения работ и вопросов, по которым предстоит получить экспертное заключение. Проект задания согласовывается начальником отдела, осуществляющего регистрацию лекарственных средств, и утверждается лицом, уполномоченным для этой цели руководителем Федеральной службы по надзору в сфере здравоохранения и социального развития. Подготовка и утверждение задания осуществляются в течение 3 рабочих дней с даты определения необходимости в проведении экспертизы.
3.3.3. Экспертная организация (экспертные организации) проводит(ят) экспертизу качества, эффективности и безопасности лекарственных средств на основании задания в сроки, определяемые заданием и общими сроками выполнения административных процедур, в рамках которых организуется проведение экспертизы. Ответственность за контроль хода проведения экспертизы несет начальник отдела, осуществляющего регистрацию лекарственных средств.
Образцы субстанций для проведения экспертизы качества с целью регистрации лекарственного средства направляются с сопроводительным письмом, в котором указывается вид контроля качества лекарственных средств, и документом, подтверждающим качество субстанции организации-производителя.
3.3.4. Приемку заключения(й) экспертной(ых) организации(й) по всем вопросам, указанным в задании, осуществляет начальник отдела, осуществляющего регистрацию лекарственных средств, и в течение 5 рабочих дней с даты приемки передает его ответственному исполнителю. Ответственный исполнитель осуществляет дальнейшее использование заключения экспертизы в соответствии с требованиями административных процедур, в связи с выполнением которых было принято решение о проведении экспертизы качества, эффективности и безопасности лекарственного средства.
3.4. Административная процедура "Организация проведения экспертизы качества при осуществлении предварительного государственного контроля лекарственных средств" выполняется в связи с поступлением от организации комплекта соответствующих документов и данных в соответствии с нижеследующим порядком (схема осуществления административной процедуры приведена в Приложении 3):
3.4.1. Документы и данные, предназначенные для организации проведения экспертизы качества при осуществлении предварительного государственного контроля лекарственных средств, поступившие от организации, регистрируются в течение 1 рабочего дня с даты получения. Комплект документов может быть направлен по почте заказным письмом (бандеролью) с описью вложения и уведомлением о вручении. Контроль ведения учета документов осуществляет начальник отдела, осуществляющего контроль производства лекарственных средств.
3.4.2. Лицо, назначенное начальником отдела, осуществляющего контроль производства лекарственных средств (ответственный исполнитель), проверяет комплектность представленных документов и данных, в состав которых должны входить:
- сопроводительное письмо;
- заверенная руководителем организации копия согласованного в установленном порядке технологического регламента производства лекарственного средства;
- копия аттестата контрольных лабораторий ОКК организации-производителя на техническую компетентность в области контроля качества производимых лекарственных средств (при наличии);
- информация об используемой субстанции (производитель, реквизиты регистрационного удостоверения, государственного стандарта качества (нормативной документации)).
Требовать от организации представления иных документов и данных не допускается. При необходимости установить достоверность сведений о государственной регистрации или о наличии утвержденного государственного стандарта качества (нормативной документации) на лекарственное средство эти данные могут быть получены ответственным исполнителем в архиве Росздравнадзора.
При отсутствии замечаний к комплектности представленных организацией документов и данных в течение 5 рабочих дней с даты регистрации комплекта документов и данных выдается разрешение на наработку трех опытно-промышленных серий лекарственного средства.
При наличии замечаний к комплектности представленных организацией документов и данных ответственный исполнитель готовит письменный отказ в проведении экспертизы качества при осуществлении предварительного государственного контроля лекарственных средств с указанием причин отказа, который подписывается руководителем Федеральной службы по надзору в сфере здравоохранения и социального развития и направляется в обратившуюся организацию.
3.4.3. На основании полученного разрешения обратившаяся организация в соответствии со своим собственным графиком осуществляет наработку трех опытно-промышленных серий лекарственного средства и по завершении письменно ставит в известность ответственного исполнителя о своей готовности предоставить образцы для осуществления предварительного государственного контроля качества лекарственного средства.
3.4.4. Ответственный исполнитель готовит проект задания на проведение экспертизы качества с указанием организации, которая будет проводить экспертизу, предельных сроков выполнения работ и вопросов, по которым предстоит получить экспертное заключение. В задании также должно быть указано, кто отвечает за отбор образцов. Проект задания согласовывается начальником отдела, осуществляющего контроль производства лекарственных средств, и утверждается лицом, уполномоченным для этой цели руководителем Федеральной службы по надзору в сфере здравоохранения и социального развития. Подготовка и утверждение задания осуществляются в течение в течение 3 рабочих дней с даты готовности обратившейся организации предоставить образцы.
3.4.5. В зависимости от обозначенного в задании на проведение экспертизы отбор образцов может производиться представителями территориальных управлений Росздравнадзора, ведомственных учреждений Росздравнадзора, экспертных организаций. Отбор образцов лекарственных средств и передача их в экспертную организацию производятся в течение 5 рабочих дней с даты утверждения задания на проведение экспертизы.
Особые требования к образцам, порядку их отбора и направления в ходе организации проведения экспертизы качества при осуществлении предварительного государственного контроля лекарственных средств:
- отбор образцов отечественных лекарственных средств осуществляется с участием представителей отдела контроля качества организации-производителя;
- образцы лекарственных средств направляются в упаковке и с маркировкой, предусмотренной государственным стандартом качества (нормативной документацией); образцы субстанций - в таре из стекла;
- образцы лекарственных средств направляются с сопроводительным письмом, в котором указывается вид контроля качества лекарственных средств, вместе с документом, подтверждающим качество лекарственного средства организации-производителя, и актом отбора образцов лекарственных средств;
- образцы лекарственных средств направляются в количестве, достаточном для проведения трех анализов в соответствии с требованиями государственного стандарта качества лекарственного средства (нормативной документации) (с учетом испытания на микробиологическую чистоту);
- образцы лекарственных средств для инъекций и глазных капель направляются с учетом испытаний показателя "механические включения", а образцы лекарственного растительного сырья - с учетом результатов радиационного контроля;
- образцы готовых лекарственных средств и образцы субстанций должны сопровождаться стандартными образцами субстанций и веществ, необходимых для проведения контроля в соответствии с государственными стандартами качества (нормативной документацией) и оригиналом или заверенной копией документа, подтверждающего качество стандартного образца организации-производителя;
- образцы лекарственных средств направляются вместе с образцом субстанции, из которой они изготовлены. Образцы субстанции направляются в количестве, достаточном для проведения двух анализов в соответствии с утвержденным государственным стандартом качества (нормативной документацией); образцы субстанций, из которых произведены лекарственные средства отечественными организациями-производителями, должны сопровождаться документами, подтверждающими качество субстанций, выданных производителем субстанции и производителем лекарственного средства по результатам входного контроля качества;
- образцы лекарственных средств, оставшиеся после проведения контроля качества лекарственных средств, хранятся не менее 6 месяцев, после чего образцы лекарственных средств, не удовлетворяющие требованиям государственного стандарта качества (нормативной документации), подлежат уничтожению в установленном порядке; образцы лекарственных средств, удовлетворяющие требованиям государственных стандартов качества (нормативной документации), возвращаются организациям-производителям по их письменной просьбе.
3.4.6. На основании полученного задания экспертная организация проводит экспертизу качества образцов в срок, определяемый заданием, но не превышающий 30 дней с даты получения образцов экспертной организацией. Ответственность за контроль хода проведения экспертизы несет начальник отдела, осуществляющего контроль производства лекарственных средств.
3.4.7. Приемку заключения экспертной организации по всем вопросам, указанным в задании, осуществляет начальник отдела, осуществляющего контроль производства лекарственных средств, и в течение 3 рабочих дней с даты приемки передает его ответственному исполнителю.
3.4.8. На основании заключения экспертизы ответственный исполнитель в течение 3 рабочих дней с даты его получения готовит проект решения по результатам экспертизы качества при осуществлении предварительного государственного контроля лекарственных средств, согласовывает его с начальником отдела, осуществляющего контроль производства лекарственных средств. Окончательное решение по результатам экспертизы качества при осуществлении предварительного государственного контроля лекарственных средств принимает руководитель Федеральной службы по надзору в сфере здравоохранения и социального развития.
3.4.9. В случае положительного решения Федеральной службы по надзору в сфере здравоохранения и социального развития по результатам предварительного государственного контроля три подвергшиеся контролю серии лекарственных средств могут быть выпущены в обращение после прохождения обязательной сертификации без повторного отбора образцов.
3.5. Административная процедура "Организация проведения экспертизы качества при осуществлении выборочного государственного контроля лекарственных средств" выполняется на основании плана выборочного контроля качества лекарственных средств, формируемого в ходе выполнения административной процедуры "Осуществление сбора и анализа информации о качестве лекарственных средств" настоящего Регламента, в соответствии с нижеследующим порядком (схема осуществления административной процедуры приведена в Приложении 4):
3.5.1. В течение 5 дней с даты наступления очередного этапа выполнения плана выборочного контроля качества лекарственных средств и на основании перечня лекарственных средств, подлежащих выборочному контролю, ответственный исполнитель, назначенный начальником отдела, осуществляющего контроль обращения лекарственных средств, готовит проект задания на проведение экспертизы качества с указанием организации, которая будет проводить экспертизу, предельных сроков выполнения работ и вопросов, по которым предстоит получить экспертное заключение. В задании также должно быть указано, кто отвечает за отбор образцов. Проект задания согласовывается начальником отдела, осуществляющего контроль обращения лекарственных средств, и утверждается лицом, уполномоченным для этой цели руководителем Федеральной службы по надзору в сфере здравоохранения и социального развития.
3.5.2. В зависимости от обозначенного в задании на проведение экспертизы отбор образцов может производиться представителями территориальных управлений Росздравнадзора, ведомственных учреждений Росздравнадзора, экспертных организаций. Отбор образцов лекарственных средств и передача их в экспертную организацию производится в течение 5 рабочих дней с даты утверждения задания на проведение экспертизы.
Особые требования к образцам, порядку их отбора и направления в ходе организации проведения экспертизы качества при осуществлении выборочного государственного контроля лекарственных средств:
- отбор образцов отечественных лекарственных средств осуществляется со складов или в аптечных организациях с участием представителей отдела контроля качества организации-производителя;
- отбор образцов зарубежных лекарственных средств проводится со складов или в аптечных организациях на территории Российской Федерации, указанных организацией, поименованной в регистрационном удостоверении на данное лекарственное средство, либо другим лицом по его поручению;
- отбор образцов лекарственных средств может проводиться при проверках организации-производителя с целью инспектирования качества лекарственных средств;
- образцы лекарственных средств направляются в упаковке, предусмотренной государственным стандартом качества (нормативной документацией); образцы субстанций - в таре из стекла. Маркировка образцов лекарственных средств должна соответствовать требованиям государственных стандартов качества (нормативной документации);
- образцы лекарственных средств направляются с сопроводительным письмом, в котором указывается вид контроля качества лекарственных средств, вместе с документом, подтверждающим качество лекарственного средства организации-производителя, и актом отбора образцов лекарственных средств;
- образцы лекарственных средств направляются в количестве, достаточном для проведения трех анализов в соответствии с требованиями государственного стандарта качества лекарственного средства (нормативной документации) (с учетом испытания на микробиологическую чистоту);
- образцы лекарственных средств для инъекций и глазных капель направляются с учетом испытаний показателя "механические включения", а образцы лекарственного растительного сырья - с учетом результатов радиационного контроля;
- образцы готовых лекарственных средств и образцы субстанций должны сопровождаться стандартными образцами субстанций и веществ, необходимых для проведения контроля в соответствии с государственными стандартами качества (нормативной документацией) и оригиналом или заверенной копией документа, подтверждающего качество стандартного образца организации-производителя;
- образцы отечественных лекарственных средств направляются с архивным образцом субстанции в количестве, достаточном для проведения двух анализов в соответствии с утвержденным государственным стандартом качества (нормативной документацией);
- образцы лекарственных средств должны сопровождаться заверенной копией сертификата соответствия лекарственного средства с протоколом анализа либо другого документа, свидетельствующего о качестве лекарственного средства;
- образцы субстанций, из которых произведены лекарственные средства отечественными организациями-производителями, должны сопровождаться документами, подтверждающими качество субстанций, выданных производителем субстанции и производителем лекарственного средства по результатам входного контроля качества;
- образцы лекарственных средств, оставшиеся после проведения государственного контроля качества лекарственных средств, хранятся не менее 6 месяцев, после чего образцы лекарственных средств, не удовлетворяющие требованиям государственного стандарта качества (нормативной документации), подлежат уничтожению в установленном порядке; образцы лекарственных средств, удовлетворяющие требованиям государственных стандартов качества (нормативной документации), возвращаются организациям-производителям по их письменной просьбе либо используются в научно-исследовательских целях.
3.5.3. На основании полученного задания экспертная организация проводит экспертизу качества образцов в срок, определяемый заданием. Ответственность за контроль хода проведения экспертизы несет начальник отдела, осуществляющего контроль обращения лекарственных средств.
3.5.4. Приемку заключения экспертной организации по всем вопросам, указанным в задании, осуществляет начальник отдела, осуществляющего контроль обращения лекарственных средств, и в течение 3 рабочих дней с даты приемки передает его ответственному исполнителю.
3.5.5. На основании заключения экспертизы ответственный исполнитель в течение 5 рабочих дней с даты его получения готовит проект решения по результатам экспертизы качества при осуществлении выборочного государственного контроля лекарственных средств, согласовывает его с начальником отдела, осуществляющего контроль обращения лекарственных средств. В том случае, если возникает необходимость прекращения обращения серии какого-либо лекарственного средства, окончательное решение об этом принимает руководитель Федеральной службы по надзору в сфере здравоохранения и социального развития.
3.6. Административная процедура "Организация проведения экспертизы качества при осуществлении повторного выборочного государственного контроля лекарственных средств" выполняется в случаях возникновения сомнений в качестве лекарственных средств у субъекта обращения лекарственных средств в связи с поступлением от него комплекта соответствующих документов и данных в соответствии с нижеследующим порядком (схема осуществления административной процедуры приведена в Приложении 5):
3.6.1. Документы и данные, содержащие сведения, дающие основания для сомнений в качестве лекарственного средства и предназначенные для проведения экспертизы качества в виде повторного выборочного государственного контроля лекарственных средств, поступившие от организации, регистрируются в течение 1 рабочего дня с даты получения. Комплект документов может быть направлен по почте заказным письмом (бандеролью) с описью вложения и уведомлением о вручении. Контроль за ведением учета документов осуществляет начальник отдела, осуществляющего контроль производства лекарственных средств.
3.6.2. Лицо, назначенное начальником отдела, осуществляющего контроль производства лекарственных средств, - ответственный исполнитель, в течение 5 рабочих дней с даты регистрации документов проверяет обоснованность проведения экспертизы качества. Надлежащим основанием считается выявление несоответствия показателей качества лекарственного средства требованиям государственного стандарта качества (нормативной документации) и несогласие с этим организации-производителя или импортера.
При отсутствии надлежащих оснований ответственный исполнитель готовит письменный отказ в проведении экспертизы качества при осуществлении повторного выборочного государственного контроля лекарственных средств с указанием оснований отказа, который подписывается руководителем Федеральной службы по надзору в сфере здравоохранения и социального развития и направляется в обратившуюся организацию.
3.6.3. При наличии надлежащих оснований ответственный исполнитель готовит проект задания на проведение экспертизы качества лекарственного средства с указанием организации, которая будет проводить экспертизу, предельных сроков выполнения работ и вопросов, по которым предстоит получить экспертное заключение. В задании также должно быть указано, кто отвечает за отбор образцов. Проект задания согласовывается начальником отдела, осуществляющего контроль производства лекарственных средств, и утверждается лицом, уполномоченным для этой цели руководителем Федеральной службы по надзору в сфере здравоохранения и социального развития. Подготовка и утверждение задания на проведение экспертизы качества при осуществлении повторного выборочного государственного контроля производятся в течение 3 рабочих дней.
3.6.4. В зависимости от обозначенного в задании на проведение экспертизы отбор образцов может производиться представителями территориальных управлений Росздравнадзора, ведомственных учреждений Росздравнадзора, экспертных организаций. Отбор образцов лекарственных средств и передача их в экспертную организацию производятся в течение 5 рабочих дней с даты утверждения задания на проведение экспертизы.
Особые требования к образцам, порядку их отбора и передачи в ходе организации проведения экспертизы качества при осуществлении повторного выборочного государственного контроля лекарственных средств:
- субъект обращения лекарственных средств на территории Российской Федерации, выявивший несоответствие лекарственных средств требованиям государственного стандарта качества (нормативной документации), представляет образцы несоответствующего лекарственного средства в количестве, достаточном для проведения необходимых анализов;
- отбор образцов осуществляется из архивных образцов лекарственных средств ОКК организации-производителя;
- образцы лекарственных средств направляются в упаковке, предусмотренной государственным стандартом качества (нормативной документацией); образцы субстанций - в таре из стекла; маркировка образцов лекарственных средств должна соответствовать требованиям государственных стандартов качества (нормативной документации);
- образцы лекарственных средств направляются с сопроводительным письмом, в котором указывается вид контроля качества лекарственных средств, вместе с документом, подтверждающим качество лекарственного средства организации-производителя и актом отбора образцов лекарственных средств;
- образцы лекарственных средств для инъекций и глазных капель направляются с учетом испытаний показателя "механические включения", а образцы лекарственного растительного сырья - с учетом результатов радиационного контроля;
- организация-производитель направляет образцы лекарственных средств в ненарушенной упаковке;
- количество образцов лекарственных средств, направляемых на повторный выборочный контроль качества по показателям "механические включения" и "радиационный контроль", определяется соответствующими государственными стандартами качества (нормативной документацией);
- образцы лекарственных средств должны сопровождаться заверенной копией сертификата соответствия лекарственного средства с протоколом анализа либо другого документа, свидетельствующего о качестве лекарственного средства;
- стандартные образцы субстанции должны сопровождаться оригиналом или заверенной копией документа, подтверждающего качество стандартного образца организации-производителя;
- образцы лекарственных средств, оставшиеся после проведения государственного контроля качества лекарственных средств, хранятся не менее 6 месяцев, после чего образцы лекарственных средств, не удовлетворяющие требованиям государственного стандарта качества (нормативной документации), подлежат уничтожению в установленном порядке; образцы лекарственных средств, удовлетворяющие требованиям государственных стандартов качества (нормативной документации), возвращаются организациям-производителям по их письменной просьбе либо используются в научно-исследовательских целях.
3.6.5. На основании полученного задания экспертная организация проводит экспертизу качества образцов в срок, определяемый заданием, но не превышающий 30 дней с даты получения образцов экспертной организацией. Ответственность за контроль хода проведения экспертизы несет начальник отдела, осуществляющего контроль производства лекарственных средств.
3.6.6. Приемку заключения экспертной организации по всем вопросам, указанным в задании, осуществляет начальник отдела, осуществляющего контроль производства лекарственных средств, и в течение 3 рабочих дней с даты приемки передает его ответственному исполнителю.
3.6.7. На основании заключения экспертизы ответственный исполнитель в течение 3 рабочих дней с даты его получения готовит проект решения по результатам экспертизы качества, согласовывает его с начальником отдела, осуществляющего контроль производства лекарственных средств. Решение по результатам экспертизы качества при осуществлении повторного выборочного государственного контроля принимает руководитель Федеральной службы по надзору в сфере здравоохранения и социального развития.
3.7. Административная процедура "Осуществление сбора и анализа информации о качестве лекарственных средств" осуществляется постоянно, в соответствии с нижеследующим порядком (схема осуществления административной процедуры приведена в Приложении 6):
3.7.1. Лица, ответственные за ведение базы данных и за контроль ведения базы данных информации о качестве лекарственных средств, по мере необходимости назначаются начальником отдела, осуществляющего сбор и анализ информации о качестве лекарственных средств, из числа сотрудников отдела.
3.7.2. Источниками документированных сведений о качестве лекарственных средств при осуществлении сбора и анализа информации являются:
- перечни серий лекарственных средств, выпущенных в обращение организациями-производителями или импортерами, направляемые в соответствии с требованиями п. 1.12 настоящего Регламента;
- результаты экспертизы качества в виде предварительного государственного контроля лекарственных средств;
- результаты экспертизы качества в виде выборочного государственного контроля лекарственных средств;
- результаты экспертизы качества в виде повторного выборочного государственного контроля лекарственных средств;
- информация о качестве лекарственных средств, полученная в ходе сбора и анализа информации о побочных эффектах применения лекарственных средств;
- информация о качестве лекарственных средств, полученная в ходе работы с обращениями частных лиц, предпринимательских, государственных и общественных организаций.
Сбор, обработка, анализ поступающих сведений, внесение изменений в базу данных информации о качестве лекарственных средств и контроль ведения базы данных должны осуществляться соответствующими ответственными лицами постоянно.
3.7.3. На основании полученных сведений о качестве лекарственных средств начальник отдела, осуществляющего сбор и анализ информации о качестве лекарственных средств, организует подготовку:
- ежемесячных отчетов о качестве лекарственных средств;
- ежегодных отчетов о качестве лекарственных средств.
3.7.4. На основании полученных сведений о качестве лекарственных средств начальник отдела, осуществляющего сбор и анализ информации о качестве лекарственных средств, организует подготовку ежеквартально обновляемого плана и перечня лекарственных средств, подлежащих выборочному государственному контролю, с учетом следующих факторов:
- частота проблем с качеством лекарственного средства;
- частота проблем с качеством лекарственных средств конкретного производителя;
- потенциальная опасность лекарственного средства (средства с высокой опасностью - стерильные лекарственные средства для парентерального введения, детские лекарственные средства, лекарственные средства, предназначенные для длительного использования);
- включение лекарственного средства в перечень дополнительного лекарственного обеспечения граждан, имеющих право на получение государственной социальной помощи.
3.7.5. По мере выявления в ходе сбора и анализа информации о качестве лекарственных средств обстоятельств, угрожающих жизни и здоровью людей при приеме лекарственных средств, ответственные лица обязаны немедленно сообщать об этом руководству Федеральной службы по надзору в сфере здравоохранения и социального развития.
3.8. Административная процедура "Осуществление сбора и анализа информации о побочных эффектах применения лекарственных средств" осуществляется постоянно, в соответствии с нижеследующим порядком (схема осуществления административной процедуры приведена в Приложении 7):
3.8.1. Лица, ответственные за ведение базы данных и за контроль ведения базы данных информации о побочных эффектах применения лекарственных средств, по мере необходимости назначаются начальником отдела, осуществляющего регистрацию лекарственных средств, из числа сотрудников отдела.
3.8.2. Источниками документированных сведений о побочных эффектах применения лекарственных средств при осуществлении сбора и анализа информации являются:
- собственные сведения Федеральной службы по надзору в сфере здравоохранения и социального развития о случаях побочных действий лекарственных средств и об особенностях взаимодействия лекарственных средств с другими средствами, которые не соответствуют сведениям, содержащимся в инструкциях по их применению;
- информация о побочных действиях лекарственных средств, полученная в ходе работы с обращениями частных лиц, предпринимательских, государственных и общественных организаций.
Сбор, обработка, анализ поступающих сведений, внесение изменений в базу данных информации о побочных эффектах применения лекарственных средств и контроль ведения базы данных должны осуществляться соответствующими ответственными лицами постоянно.
3.8.3. На основании полученных сведений о побочных эффектах применения лекарственных средств начальник отдела, отвечающего регистрацию лекарственных средств, организует подготовку:
- ежемесячных отчетов о безопасности лекарственных средств;
- ежегодных отчетов о безопасности лекарственных средств.
3.8.4. По мере выявления в ходе сбора и анализа информации о побочных эффектах применения лекарственных средств обстоятельств, угрожающих жизни и здоровью людей, ответственные лица обязаны немедленно сообщать об этом руководству Федеральной службы по надзору в сфере здравоохранения и социального развития.
ФЕДЕРАЛЬНЫЙ ЗАКОН
О ТЕХНИЧЕСКОМ РЕГУЛИРОВАНИИ
(в ред. Федеральных законов от 28.12.2013 N 396-ФЗ)
(извлечение)
Глава 1. ОБЩИЕ ПОЛОЖЕНИЯ
Статья 1. Сфера применения настоящего Федерального закона
1. Настоящий Федеральный закон регулирует отношения, возникающие при:
разработке, принятии, применении и исполнении обязательных требований к продукции, в том числе зданиям и сооружениям (далее - продукция), или к продукции и связанным с требованиями к продукции процессам проектирования (включая изыскания), производства, строительства, монтажа, наладки, эксплуатации, хранения, перевозки, реализации и утилизации;
(в ред. Федеральных законов от 01.05.2007 N 65-ФЗ, от 21.07.2011 N 255-ФЗ)
разработке, принятии, применении и исполнении на добровольной основе требований к продукции, процессам проектирования (включая изыскания), производства, строительства, монтажа, наладки, эксплуатации, хранения, перевозки, реализации и утилизации, выполнению работ или оказанию услуг;
(в ред. Федерального закона от 01.05.2007 N 65-ФЗ)
оценке соответствия.
Настоящий Федеральный закон также определяет права и обязанности участников регулируемых настоящим Федеральным законом отношений.
2. Требования к функционированию единой сети связи Российской Федерации, связанные с обеспечением целостности, устойчивости функционирования указанной сети связи и ее безопасности, отношения, связанные с обеспечением целостности единой сети связи Российской Федерации и использованием радиочастотного спектра, соответственно устанавливаются и регулируются законодательством Российской Федерации в области связи.
(в ред. Федерального закона от 21.07.2011 N 255-ФЗ)
3. Действие настоящего Федерального закона не распространяется на социально-экономические, организационные, санитарно-гигиенические, лечебно-профилактические, реабилитационные меры в области охраны труда, федеральные государственные образовательные стандарты, положения (стандарты) о бухгалтерском учете и правила (стандарты) аудиторской деятельности, стандарты эмиссии ценных бумаг и проспектов эмиссии ценных бумаг, стандарты оценочной деятельности, стандарты распространения, предоставления или раскрытия информации, минимальные социальные стандарты, стандарты предоставления государственных и муниципальных услуг, профессиональные стандарты.
(в ред. Федеральных законов от 01.05.2007 N 65-ФЗ, от 01.12.2007 N 309-ФЗ, от 21.07.2011 N 255-ФЗ, от 03.12.2012 N 236-ФЗ)
4. Настоящий Федеральный закон не регулирует отношения, связанные с разработкой, принятием, применением и исполнением санитарно-эпидемиологических требований, требований в области охраны окружающей среды, требований в области охраны труда, требований к безопасному использованию атомной энергии, в том числе требований безопасности объектов использования атомной энергии, требований безопасности деятельности в области использования атомной энергии, требований к осуществлению деятельности в области промышленной безопасности, безопасности технологических процессов на опасных производственных объектах, требований к обеспечению надежности и безопасности электроэнергетических систем и объектов электроэнергетики, требований к обеспечению безопасности космической деятельности, за исключением случаев разработки, принятия, применения и исполнения таких требований к продукции или к продукции и связанным с требованиями к продукции процессам проектирования (включая изыскания), производства, строительства, монтажа, наладки, эксплуатации, хранения, перевозки, реализации и утилизации.
(в ред. Федеральных законов от 21.07.2011 N 255-ФЗ, от 30.11.2011 N 347-ФЗ).
Статья 2. Основные понятия
Для целей настоящего Федерального закона используются следующие основные понятия:
аккредитация - официальное признание органом по аккредитации компетентности физического или юридического лица выполнять работы в определенной области оценки соответствия;
безопасность продукции и связанных с ней процессов производства, эксплуатации, хранения, перевозки, реализации и утилизации (далее - безопасность) - состояние, при котором отсутствует недопустимый риск, связанный с причинением вреда жизни или здоровью граждан, имуществу физических или юридических лиц, государственному или муниципальному имуществу, окружающей среде, жизни или здоровью животных и растений;
(в ред. Федерального закона от 21.07.2011 N 255-ФЗ)
ветеринарно-санитарные и фитосанитарные меры - обязательные для исполнения требования и процедуры, устанавливаемые в целях защиты от рисков, возникающих в связи с проникновением, закреплением или распространением вредных организмов, заболеваний, переносчиков болезней или болезнетворных организмов, в том числе в случае переноса или распространения их животными и (или) растениями, с продукцией, грузами, материалами, транспортными средствами, с наличием добавок, загрязняющих веществ, токсинов, вредителей, сорных растений, болезнетворных организмов, в том числе с пищевыми продуктами или кормами, а также обязательные для исполнения требования и процедуры, устанавливаемые в целях предотвращения иного связанного с распространением вредных организмов ущерба;
декларирование соответствия - форма подтверждения соответствия продукции требованиям технических регламентов;
декларация о соответствии - документ, удостоверяющий соответствие выпускаемой в обращение продукции требованиям технических регламентов;
заявитель - физическое или юридическое лицо, которое для подтверждения соответствия принимает декларацию о соответствии или обращается за получением сертификата соответствия, получает сертификат соответствия;
(в ред. Федерального закона от 01.05.2007 N 65-ФЗ)
знак обращения на рынке - обозначение, служащее для информирования приобретателей, в том числе потребителей, о соответствии выпускаемой в обращение продукции требованиям технических регламентов;
(в ред. Федерального закона от 21.07.2011 N 255-ФЗ)
знак соответствия - обозначение, служащее для информирования приобретателей, в том числе потребителей, о соответствии объекта сертификации требованиям системы добровольной сертификации или национальному стандарту;
(в ред. Федерального закона от 21.07.2011 N 255-ФЗ)
идентификация продукции - установление тождественности характеристик продукции ее существенным признакам;
контроль (надзор) за соблюдением требований технических регламентов - проверка выполнения юридическим лицом или индивидуальным предпринимателем требований технических регламентов к продукции или к продукции и связанным с требованиями к продукции процессам проектирования (включая изыскания), производства, строительства, монтажа, наладки, эксплуатации, хранения, перевозки, реализации и утилизации и принятие мер по результатам проверки;
(в ред. Федеральных законов от 01.05.2007 N 65-ФЗ, от 21.07.2011 N 255-ФЗ)
международный стандарт - стандарт, принятый международной организацией;
национальный стандарт - стандарт, утвержденный национальным органом Российской Федерации по стандартизации;
орган по сертификации - юридическое лицо или индивидуальный предприниматель, аккредитованные в установленном порядке для выполнения работ по сертификации;
оценка соответствия - прямое или косвенное определение соблюдения требований, предъявляемых к объекту;
подтверждение соответствия - документальное удостоверение соответствия продукции или иных объектов, процессов проектирования (включая изыскания), производства, строительства, монтажа, наладки, эксплуатации, хранения, перевозки, реализации и утилизации, выполнения работ или оказания услуг требованиям технических регламентов, положениям стандартов, сводов правил или условиям договоров;
(в ред. Федерального закона от 01.05.2007 N 65-ФЗ)
продукция - результат деятельности, представленный в материально-вещественной форме и предназначенный для дальнейшего использования в хозяйственных и иных целях;
риск - вероятность причинения вреда жизни или здоровью граждан, имуществу физических или юридических лиц, государственному или муниципальному имуществу, окружающей среде, жизни или здоровью животных и растений с учетом тяжести этого вреда;
сертификация - форма осуществляемого органом по сертификации подтверждения соответствия объектов требованиям технических регламентов, положениям стандартов, сводов правил или условиям договоров;
(в ред. Федерального закона от 01.05.2007 N 65-ФЗ)
сертификат соответствия - документ, удостоверяющий соответствие объекта требованиям технических регламентов, положениям стандартов, сводов правил или условиям договоров;
(в ред. Федерального закона от 01.05.2007 N 65-ФЗ)
система сертификации - совокупность правил выполнения работ по сертификации, ее участников и правил функционирования системы сертификации в целом;
стандарт - документ, в котором в целях добровольного многократного использования устанавливаются характеристики продукции, правила осуществления и характеристики процессов проектирования (включая изыскания), производства, строительства, монтажа, наладки, эксплуатации, хранения, перевозки, реализации и утилизации, выполнения работ или оказания услуг. Стандарт также может содержать правила и методы исследований (испытаний) и измерений, правила отбора образцов, требования к терминологии, символике, упаковке, маркировке или этикеткам и правилам их нанесения;
(в ред. Федерального закона от 01.05.2007 N 65-ФЗ)
стандартизация - деятельность по установлению правил и характеристик в целях их добровольного многократного использования, направленная на достижение упорядоченности в сферах производства и обращения продукции и повышение конкурентоспособности продукции, работ или услуг;
техническое регулирование - правовое регулирование отношений в области установления, применения и исполнения обязательных требований к продукции или к продукции и связанным с требованиями к продукции процессам проектирования (включая изыскания), производства, строительства, монтажа, наладки, эксплуатации, хранения, перевозки, реализации и утилизации, а также в области установления и применения на добровольной основе требований к продукции, процессам проектирования (включая изыскания), производства, строительства, монтажа, наладки, эксплуатации, хранения, перевозки, реализации и утилизации, выполнению работ или оказанию услуг и правовое регулирование отношений в области оценки соответствия;
(в ред. Федеральных законов от 01.05.2007 N 65-ФЗ, от 21.07.2011 N 255-ФЗ)
технический регламент - документ, который принят международным договором Российской Федерации, подлежащим ратификации в порядке, установленном законодательством Российской Федерации, или в соответствии с международным договором Российской Федерации, ратифицированным в порядке, установленном законодательством Российской Федерации, или федеральным законом, или указом Президента Российской Федерации, или постановлением Правительства Российской Федерации, или нормативным правовым актом федерального органа исполнительной власти по техническому регулированию и устанавливает обязательные для применения и исполнения требования к объектам технического регулирования (продукции или к продукции и связанным с требованиями к продукции процессам проектирования (включая изыскания), производства, строительства, монтажа, наладки, эксплуатации, хранения, перевозки, реализации и утилизации);
(в ред. Федерального закона от 21.07.2011 N 255-ФЗ)
форма подтверждения соответствия - определенный порядок документального удостоверения соответствия продукции или иных объектов, процессов проектирования (включая изыскания), производства, строительства, монтажа, наладки, эксплуатации, хранения, перевозки, реализации и утилизации, выполнения работ или оказания услуг требованиям технических регламентов, положениям стандартов или условиям договоров;
(в ред. Федерального закона от 01.05.2007 N 65-ФЗ)
схема подтверждения соответствия - перечень действий участников подтверждения соответствия, результаты которых рассматриваются ими в качестве доказательств соответствия продукции и иных объектов установленным требованиям;
(абзац введен Федеральным законом от 01.05.2007 N 65-ФЗ)
свод правил - документ в области стандартизации, в котором содержатся технические правила и (или) описание процессов проектирования (включая изыскания), производства, строительства, монтажа, наладки, эксплуатации, хранения, перевозки, реализации и утилизации продукции и который применяется на добровольной основе в целях соблюдения требований технических регламентов;
(абзац введен Федеральным законом от 01.05.2007 N 65-ФЗ, в ред. Федерального закона от 18.07.2009 N 189-ФЗ)
региональная организация по стандартизации - организация, членами (участниками) которой являются национальные органы (организации) по стандартизации государств, входящих в один географический регион мира и (или) группу стран, находящихся в соответствии с международными договорами в процессе экономической интеграции;
(абзац введен Федеральным законом от 30.12.2009 N 385-ФЗ)
стандарт иностранного государства - стандарт, принятый национальным (компетентным) органом (организацией) по стандартизации иностранного государства;
(абзац введен Федеральным законом от 30.12.2009 N 385-ФЗ)
региональный стандарт - стандарт, принятый региональной организацией по стандартизации;
(абзац введен Федеральным законом от 30.12.2009 N 385-ФЗ)
свод правил иностранного государства - свод правил, принятый компетентным органом иностранного государства;
(абзац введен Федеральным законом от 30.12.2009 N 385-ФЗ)
региональный свод правил - свод правил, принятый региональной организацией по стандартизации;
(абзац введен Федеральным законом от 30.12.2009 N 385-ФЗ)
предварительный национальный стандарт - документ в области стандартизации, который утвержден национальным органом Российской Федерации по стандартизации и срок действия которого ограничен;
(абзац введен Федеральным законом от 21.07.2011 N 255-ФЗ)
аттестат аккредитации - документ, удостоверяющий аккредитацию лица в качестве органа по сертификации или испытательной лаборатории (центра) в определенной области аккредитации;
(абзац введен Федеральным законом от 21.07.2011 N 255-ФЗ)
область аккредитации - сфера деятельности органа по сертификации, испытательной лаборатории (центра), определяемая при их аккредитации;
(абзац введен Федеральным законом от 21.07.2011 N 255-ФЗ)
впервые выпускаемая в обращение продукция - продукция, которая ранее не находилась в обращении на территории Российской Федерации либо которая ранее выпускалась в обращение и свойства или характеристики которой были впоследствии изменены.
(абзац введен Федеральным законом от 21.07.2011 N 255-ФЗ)
Статья 3. Принципы технического регулирования
Техническое регулирование осуществляется в соответствии с принципами:
применения единых правил установления требований к продукции или к продукции и связанным с требованиями к продукции процессам проектирования (включая изыскания), производства, строительства, монтажа, наладки, эксплуатации, хранения, перевозки, реализации и утилизации, выполнению работ или оказанию услуг;
(в ред. Федеральных законов от 01.05.2007 N 65-ФЗ, от 21.07.2011 N 255-ФЗ)
соответствия технического регулирования уровню развития национальной экономики, развития материально-технической базы, а также уровню научно-технического развития;
независимости органов по аккредитации, органов по сертификации от изготовителей, продавцов, исполнителей и приобретателей, в том числе потребителей;
(в ред. Федерального закона от 21.07.2011 N 255-ФЗ)
единой системы и правил аккредитации;
единства правил и методов исследований (испытаний) и измерений при проведении процедур обязательной оценки соответствия;
единства применения требований технических регламентов независимо от видов или особенностей сделок;
недопустимости ограничения конкуренции при осуществлении аккредитации и сертификации;
недопустимости совмещения одним органом полномочий по государственному контролю (надзору), за исключением осуществления контроля за деятельностью аккредитованных лиц, с полномочиями по аккредитации или сертификации;
(в ред. Федерального закона от 21.07.2011 N 255-ФЗ)
недопустимости совмещения одним органом полномочий по аккредитации и сертификации;
(в ред. Федерального закона от 21.07.2011 N 255-ФЗ)
недопустимости внебюджетного финансирования государственного контроля (надзора) за соблюдением требований технических регламентов;
недопустимости одновременного возложения одних и тех же полномочий на два и более органа государственного контроля (надзора) за соблюдением требований технических регламентов.
(абзац введен Федеральным законом от 01.05.2007 N 65-ФЗ)
Статья 4. Законодательство Российской Федерации о техническом регулировании
1. Законодательство Российской Федерации о техническом регулировании состоит из настоящего Федерального закона, принимаемых в соответствии с ним федеральных законов и иных нормативных правовых актов Российской Федерации.
2. Положения федеральных законов и иных нормативных правовых актов Российской Федерации, касающиеся сферы применения настоящего Федерального закона (в том числе прямо или косвенно предусматривающие осуществление контроля (надзора) за соблюдением требований технических регламентов), применяются в части, не противоречащей настоящему Федеральному закону.
3. Федеральные органы исполнительной власти вправе издавать в сфере технического регулирования акты только рекомендательного характера, за исключением случаев, установленных статьями 5 и 9.1 настоящего Федерального закона.
(в ред. Федерального закона от 30.12.2009 N 385-ФЗ)
4. Если международным договором Российской Федерации в сфере технического регулирования установлены иные правила, чем те, которые предусмотрены настоящим Федеральным законом, применяются правила международного договора, а в случаях, если из международного договора следует, что для его применения требуется издание внутригосударственного акта, применяются правила международного договора и принятое на его основе законодательство Российской Федерации.
Глава 2. ТЕХНИЧЕСКИЕ РЕГЛАМЕНТЫ
Статья 6. Цели принятия технических регламентов
1. Технические регламенты принимаются в целях:
защиты жизни или здоровья граждан, имущества физических или юридических лиц, государственного или муниципального имущества;
охраны окружающей среды, жизни или здоровья животных и растений;
предупреждения действий, вводящих в заблуждение приобретателей, в том числе потребителей;
(в ред. Федерального закона от 21.07.2011 N 255-ФЗ)
обеспечения энергетической эффективности и ресурсосбережения.
(абзац введен Федеральным законом от 18.07.2009 N 189-ФЗ, в ред. Федерального закона от 21.07.2011 N 255-ФЗ)
2. Принятие технических регламентов в иных целях не допускается.
Статья 7. Содержание и применение технических регламентов
1. Технические регламенты с учетом степени риска причинения вреда устанавливают минимально необходимые требования, обеспечивающие:
безопасность излучений;
биологическую безопасность;
взрывобезопасность;
механическую безопасность;
пожарную безопасность;
безопасность продукции (технических устройств, применяемых на опасном производственном объекте);
(в ред. Федерального закона от 21.07.2011 N 255-ФЗ)
термическую безопасность;
химическую безопасность;
электрическую безопасность;
радиационную безопасность населения;
(в ред. Федеральных законов от 21.07.2011 N 255-ФЗ, от 30.11.2011 N 347-ФЗ)
электромагнитную совместимость в части обеспечения безопасности работы приборов и оборудования;
единство измерений;
другие виды безопасности в целях, соответствующих пункту 1 статьи 6 настоящего Федерального закона.
(абзац введен Федеральным законом от 01.05.2007 N 65-ФЗ)
2. Требования технических регламентов не могут служить препятствием осуществлению предпринимательской деятельности в большей степени, чем это минимально необходимо для выполнения целей, указанных в пункте 1 статьи 6 настоящего Федерального закона.
3. Технический регламент должен содержать перечень и (или) описание объектов технического регулирования, требования к этим объектам и правила их идентификации в целях применения технического регламента. Технический регламент должен содержать правила и формы оценки соответствия (в том числе в техническом регламенте могут содержаться схемы подтверждения соответствия, порядок продления срока действия выданного сертификата соответствия), определяемые с учетом степени риска, предельные сроки оценки соответствия в отношении каждого объекта технического регулирования и (или) требования к терминологии, упаковке, маркировке или этикеткам и правилам их нанесения. Технический регламент должен содержать требования энергетической эффективности и ресурсосбережения.
(в ред. Федеральных законов от 01.05.2007 N 65-ФЗ, от 18.07.2009 N 189-ФЗ, от 21.07.2011 N 255-ФЗ)
Оценка соответствия проводится в формах государственного контроля (надзора), испытания, регистрации, подтверждения соответствия, приемки и ввода в эксплуатацию объекта, строительство которого закончено, и в иной форме.
(в ред. Федерального закона от 21.07.2011 N 255-ФЗ)
Содержащиеся в технических регламентах обязательные требования к продукции или к продукции и связанным с требованиями к продукции процессам проектирования (включая изыскания), производства, строительства, монтажа, наладки, эксплуатации, хранения, перевозки, реализации и утилизации, правилам и формам оценки соответствия, правила идентификации, требования к терминологии, упаковке, маркировке или этикеткам и правилам их нанесения имеют прямое действие на всей территории Российской Федерации и могут быть изменены только путем внесения изменений и дополнений в соответствующий технический регламент.
(в ред. Федеральных законов от 01.05.2007 N 65-ФЗ, от 21.07.2011 N 255-ФЗ)
Не включенные в технические регламенты требования к продукции или к продукции и связанным с требованиями к продукции процессам проектирования (включая изыскания), производства, строительства, монтажа, наладки, эксплуатации, хранения, перевозки, реализации и утилизации, правилам и формам оценки соответствия, правила идентификации, требования к терминологии, упаковке, маркировке или этикеткам и правилам их нанесения не могут носить обязательный характер.
(в ред. Федеральных законов от 01.05.2007 N 65-ФЗ, от 21.07.2011 N 255-ФЗ)
4. Технический регламент должен содержать обобщенные и (или) конкретные требования к характеристикам продукции или к продукции и связанным с требованиями к продукции процессам проектирования (включая изыскания), производства, строительства, монтажа, наладки, эксплуатации, хранения, перевозки, реализации и утилизации, но не должен содержать требования к конструкции и исполнению, за исключением случаев, если из-за отсутствия требований к конструкции и исполнению с учетом степени риска причинения вреда не обеспечивается достижение указанных в пункте 1 статьи 6 настоящего Федерального закона целей принятия технического регламента.
(в ред. Федеральных законов от 01.05.2007 N 65-ФЗ, от 21.07.2011 N 255-ФЗ)
5. В технических регламентах с учетом степени риска причинения вреда могут содержаться специальные требования к продукции или к продукции и связанным с требованиями к продукции процессам проектирования (включая изыскания), производства, строительства, монтажа, наладки, эксплуатации, хранения, перевозки, реализации и утилизации, требования к терминологии, упаковке, маркировке или этикеткам и правилам их нанесения, обеспечивающие защиту отдельных категорий граждан (несовершеннолетних, беременных женщин, кормящих матерей, инвалидов).
(в ред. Федеральных законов от 01.05.2007 N 65-ФЗ, от 21.07.2011 N 255-ФЗ)
6. Технические регламенты применяются одинаковым образом и в равной мере независимо от вида нормативного правового акта, которым они приняты, страны и (или) места происхождения продукции или осуществления связанных с требованиями к продукции процессов проектирования (включая изыскания), производства, строительства, монтажа, наладки, эксплуатации, хранения, перевозки, реализации и утилизации, видов или особенностей сделок и (или) физических и (или) юридических лиц, являющихся изготовителями, исполнителями, продавцами, приобретателями, в том числе потребителями, с учетом положений пункта 9 настоящей статьи.
(в ред. Федеральных законов от 01.05.2007 N 65-ФЗ, от 21.07.2011 N 255-ФЗ)
7. Технический регламент не может содержать требования к продукции, причиняющей вред жизни или здоровью граждан, накапливаемый при длительном использовании этой продукции и зависящий от других факторов, не позволяющих определить степень допустимого риска. В этих случаях технический регламент может содержать требование, касающееся информирования приобретателя, в том числе потребителя, о возможном вреде и о факторах, от которых он зависит.
(в ред. Федерального закона от 21.07.2011 N 255-ФЗ)
8. Международные стандарты должны использоваться полностью или частично в качестве основы для разработки проектов технических регламентов, за исключением случаев, если международные стандарты или их разделы были бы неэффективными или не подходящими для достижения установленных статьей 6 настоящего Федерального закона целей, в том числе вследствие климатических и географических особенностей Российской Федерации, технических и (или) технологических особенностей.
(в ред. Федерального закона от 18.07.2009 N 189-ФЗ)
Национальные стандарты могут использоваться полностью или частично в качестве основы для разработки проектов технических регламентов.
(п. 8 в ред. Федерального закона от 01.05.2007 N 65-ФЗ)
9. Технический регламент может содержать специальные требования к продукции или к продукции и связанным с требованиями к продукции процессам проектирования (включая изыскания), производства, строительства, монтажа, наладки, эксплуатации, хранения, перевозки, реализации и утилизации, терминологии, упаковке, маркировке или этикеткам и правилам их нанесения, применяемые в отдельных местах происхождения продукции, если отсутствие таких требований в силу климатических и географических особенностей приведет к недостижению целей, указанных в пункте 1 статьи 6 настоящего Федерального закона.
(в ред. Федеральных законов от 01.05.2007 N 65-ФЗ, от 21.07.2011 N 255-ФЗ).
Технические регламенты устанавливают также минимально необходимые ветеринарно-санитарные и фитосанитарные меры в отношении продукции, происходящей из отдельных стран и (или) мест, в том числе ограничения ввоза, использования, хранения, перевозки, реализации и утилизации, обеспечивающие биологическую безопасность (независимо от способов обеспечения безопасности, использованных изготовителем).
Ветеринарно-санитарными и фитосанитарными мерами могут предусматриваться требования к продукции, методам ее обработки и производства, процедурам испытания продукции, инспектирования, подтверждения соответствия, карантинные правила, в том числе требования, связанные с перевозкой животных и растений, необходимых для обеспечения жизни или здоровья животных и растений во время их перевозки материалов, а также методы и процедуры отбора проб, методы исследования и оценки риска и иные содержащиеся в технических регламентах требования.
Ветеринарно-санитарные и фитосанитарные меры разрабатываются и применяются на основе научных данных, а также с учетом соответствующих международных стандартов, рекомендаций и других документов международных организаций в целях соблюдения необходимого уровня ветеринарно-санитарной и фитосанитарной защиты, который определяется с учетом степени фактического научно обоснованного риска. При оценке степени риска могут приниматься во внимание положения международных стандартов, рекомендации международных организаций, участником которых является Российская Федерация, распространенность заболеваний и вредителей, а также применяемые поставщиками меры по борьбе с заболеваниями и вредителями, экологические условия, экономические последствия, связанные с возможным причинением вреда, размеры расходов на предотвращение причинения вреда.
В случае, если безотлагательное применение ветеринарно-санитарных и фитосанитарных мер необходимо для достижения целей ветеринарно-санитарной и фитосанитарной защиты, а соответствующее научное обоснование является недостаточным или не может быть получено в необходимые сроки, ветеринарно-санитарные или фитосанитарные меры, предусмотренные техническими регламентами в отношении определенных видов продукции, могут быть применены на основе имеющейся информации, в том числе информации, полученной от соответствующих международных организаций, властей иностранных государств, информации о применяемых другими государствами соответствующих мерах или иной информации. До принятия соответствующих технических регламентов в случае, установленном настоящим абзацем, ветеринарно-санитарные и фитосанитарные меры действуют в соответствии с законодательством Российской Федерации.
(в ред. Федерального закона от 21.07.2011 N 255-ФЗ)
Ветеринарно-санитарные и фитосанитарные меры должны применяться с учетом соответствующих экономических факторов - потенциального ущерба от уменьшения объема производства продукции или ее продаж в случае проникновения, закрепления или распространения какого-либо вредителя или заболевания, расходов на борьбу с ними или их ликвидацию, эффективности применения альтернативных мер по ограничению рисков, а также необходимости сведения к минимуму воздействия вредителя или заболевания на окружающую среду, производство и обращение продукции.
10. Технический регламент, принимаемый федеральным законом, постановлением Правительства Российской Федерации или нормативным правовым актом федерального органа исполнительной власти по техническому регулированию, вступает в силу не ранее чем через шесть месяцев со дня его официального опубликования.
(п. 10 в ред. Федерального закона от 30.12.2009 N 385-ФЗ)
11. Правительством Российской Федерации или в случае, предусмотренном статьей 9.1 настоящего Федерального закона, федеральным органом исполнительной власти по техническому регулированию до дня вступления в силу технического регламента утверждается в соответствии с требованиями законодательства Российской Федерации в области обеспечения единства измерений перечень документов в области стандартизации, содержащих правила и методы исследований (испытаний) и измерений, в том числе правила отбора образцов, необходимые для применения и исполнения принятого технического регламента и осуществления оценки соответствия. В случае отсутствия указанных документов в области стандартизации применительно к отдельным требованиям технического регламента или объектам технического регулирования Правительством Российской Федерации или в случае, предусмотренном статьей 9.1 настоящего Федерального закона, федеральным органом исполнительной власти по техническому регулированию до дня вступления в силу технического регламента утверждаются в соответствии с требованиями законодательства Российской Федерации в области обеспечения единства измерений правила и методы исследований (испытаний) и измерений, в том числе правила отбора образцов, необходимые для применения и исполнения принятого технического регламента и осуществления оценки соответствия. Проекты указанных правил и методов разрабатываются федеральными органами исполнительной власти в соответствии с их компетенцией или в случае, предусмотренном статьей 9.1 настоящего Федерального закона, федеральным органом исполнительной власти по техническому регулированию с использованием документов в области стандартизации, опубликовываются в печатном издании федерального органа исполнительной власти по техническому регулированию и размещаются в информационной системе общего пользования в электронно-цифровой форме не позднее чем за тридцать дней до дня утверждения указанных правил и методов.
(в ред. Федерального закона от 30.12.2009 N 385-ФЗ)
Указанные правила не могут служить препятствием осуществлению предпринимательской деятельности в большей степени, чем это минимально необходимо для выполнения целей, указанных в пункте 1 статьи 6 настоящего Федерального закона.
(п. 11 в ред. Федерального закона от 01.05.2007 N 65-ФЗ)
12. Правительство Российской Федерации разрабатывает предложения об обеспечении соответствия технического регулирования интересам национальной экономики, уровню развития материально-технической базы и уровню научно-технического развития, а также международным нормам и правилам.
(в ред. Федеральных законов от 01.05.2007 N 65-ФЗ, от 21.07.2011 N 255-ФЗ)
Уполномоченным Правительством Российской Федерации федеральным органом исполнительной власти организуются постоянные учет и анализ всех случаев причинения вреда вследствие нарушения требований технических регламентов жизни или здоровью граждан, имуществу физических или юридических лиц, государственному или муниципальному имуществу, окружающей среде, жизни или здоровью животных и растений с учетом тяжести этого вреда, а также организуется информирование приобретателей, в том числе потребителей, изготовителей и продавцов о ситуации в области соблюдения требований технических регламентов.
(в ред. Федеральных законов от 23.07.2008 N 160-ФЗ, от 21.07.2011 N 255-ФЗ).
Статья 9. Порядок разработки, принятия, изменения и отмены технического регламента.
1. Технический регламент может быть принят международным договором Российской Федерации, подлежащим ратификации в порядке, установленном законодательством Российской Федерации, или в соответствии с международным договором Российской Федерации, ратифицированным в порядке, установленном законодательством Российской Федерации. Такие технические регламенты разрабатываются, принимаются и отменяются в порядке, принятом в соответствии с международным договором Российской Федерации, ратифицированным в порядке, установленном законодательством Российской Федерации.
До вступления в силу технического регламента, принятого международным договором Российской Федерации, подлежащим ратификации в порядке, установленном законодательством Российской Федерации, или в соответствии с международным договором Российской Федерации, ратифицированным в порядке, установленном законодательством Российской Федерации, технический регламент может быть принят федеральным законом, или указом Президента Российской Федерации, или постановлением Правительства Российской Федерации, или нормативным правовым актом федерального органа исполнительной власти по техническому регулированию в соответствии с положениями настоящего Федерального закона.
Технический регламент, разработанный в порядке, установленном настоящей статьей, принимается федеральным законом или постановлением Правительства Российской Федерации в порядке, установленном соответственно для принятия федеральных законов и постановлений Правительства Российской Федерации, в соответствии с положениями настоящего Федерального закона.
(п. 1 в ред. Федерального закона от 21.07.2011 N 255-ФЗ)
2. Разработчиком проекта технического регламента может быть любое лицо.
3. О разработке проекта технического регламента должно быть опубликовано уведомление в печатном издании федерального органа исполнительной власти по техническому регулированию и в информационной системе общего пользования в электронно-цифровой форме.
Уведомление о разработке проекта технического регламента должно содержать информацию о том, в отношении какой продукции или каких связанных с требованиями к ней процессов проектирования (включая изыскания), производства, строительства, монтажа, наладки, эксплуатации, хранения, перевозки, реализации и утилизации будут устанавливаться разрабатываемые требования, с кратким изложением цели этого технического регламента, обоснованием необходимости его разработки и указанием тех разрабатываемых требований, которые отличаются от положений соответствующих международных стандартов или обязательных требований, действующих на территории Российской Федерации в момент разработки проекта данного технического регламента, и информацию о способе ознакомления с проектом технического регламента, наименование или фамилию, имя, отчество разработчика проекта данного технического регламента, почтовый адрес и при наличии адрес электронной почты, по которым должен осуществляться прием в письменной форме замечаний заинтересованных лиц.
(в ред. Федерального закона от 01.05.2007 N 65-ФЗ)
4. С момента опубликования уведомления о разработке проекта технического регламента соответствующий проект технического регламента должен быть доступен заинтересованным лицам для ознакомления. Разработчик обязан по требованию заинтересованного лица предоставить ему копию проекта технического регламента. Плата, взимаемая за предоставление данной копии, не может превышать затраты на ее изготовление.
Разработчик дорабатывает проект технического регламента с учетом полученных в письменной форме замечаний заинтересованных лиц, проводит публичное обсуждение проекта технического регламента и составляет перечень полученных в письменной форме замечаний заинтересованных лиц с кратким изложением содержания данных замечаний и результатов их обсуждения.
Разработчик обязан сохранять полученные в письменной форме замечания заинтересованных лиц до дня вступления в силу принимаемого соответствующим нормативным правовым актом технического регламента и предоставлять их депутатам Государственной Думы, представителям федеральных органов исполнительной власти и указанным в пункте 9 настоящей статьи экспертным комиссиям по техническому регулированию по их запросам.
Срок публичного обсуждения проекта технического регламента со дня опубликования уведомления о разработке проекта технического регламента до дня опубликования уведомления о завершении публичного обсуждения не может быть менее чем два месяца.
5. Уведомление о завершении публичного обсуждения проекта технического регламента должно быть опубликовано в печатном издании федерального органа исполнительной власти по техническому регулированию и в информационной системе общего пользования в электронно-цифровой форме.
Уведомление о завершении публичного обсуждения проекта технического регламента должно включать в себя информацию о способе ознакомления с проектом технического регламента и перечнем полученных в письменной форме замечаний заинтересованных лиц, а также наименование или фамилию, имя, отчество разработчика проекта технического регламента, почтовый адрес и при наличии адрес электронной почты, по которым с разработчиком может быть осуществлена связь.
Со дня опубликования уведомления о завершении публичного обсуждения проекта технического регламента доработанный проект технического регламента и перечень полученных в письменной форме замечаний заинтересованных лиц должны быть доступны заинтересованным лицам для ознакомления.
6. Федеральный орган исполнительной власти по техническому регулированию обязан опубликовывать в своем печатном издании уведомления о разработке проекта технического регламента и завершении публичного обсуждения этого проекта в течение десяти дней с момента оплаты опубликования уведомлений. Порядок опубликования уведомлений и размер платы за их опубликование устанавливаются Правительством Российской Федерации.
7. Внесение субъектом права законодательной инициативы проекта федерального закона о техническом регламенте в Государственную Думу осуществляется при наличии следующих документов:
обоснование необходимости принятия федерального закона о техническом регламенте с указанием тех требований, которые отличаются от положений соответствующих международных стандартов или обязательных требований, действующих на территории Российской Федерации в момент разработки проекта технического регламента;
финансово-экономическое обоснование принятия федерального закона о техническом регламенте;
документы, подтверждающие опубликование уведомления о разработке проекта технического регламента в соответствии с пунктом 3 настоящей статьи;
документы, подтверждающие опубликование уведомления о завершении публичного обсуждения проекта технического регламента в соответствии с пунктом 5 настоящей статьи;
перечень полученных в письменной форме замечаний заинтересованных лиц, указанный в пункте 4 настоящей статьи.
Внесенный в Государственную Думу проект федерального закона о техническом регламенте с приложением документов, указанных в настоящем пункте, направляется Государственной Думой в Правительство Российской Федерации. На проект федерального закона о техническом регламенте Правительство Российской Федерации в течение девяноста дней направляет в Государственную Думу отзыв, подготовленный с учетом заключения экспертной комиссии по техническому регулированию. Проект федерального закона о техническом регламенте может быть рассмотрен Государственной Думой в первом чтении без отзыва Правительства Российской Федерации в случае, если отзыв Правительства Российской Федерации не был представлен в Государственную Думу в указанный срок.
(в ред. Федерального закона от 01.05.2007 N 65-ФЗ)
8. Проект федерального закона о техническом регламенте, принятый Государственной Думой в первом чтении, публикуется в печатном издании федерального органа исполнительной власти по техническому регулированию и в информационной системе общего пользования в электронно-цифровой форме.
Поправки к принятому в первом чтении проекту федерального закона о техническом регламенте после окончания срока их подачи публикуются в информационной системе общего пользования в электронно-цифровой форме не позднее чем за месяц до рассмотрения Государственной Думой проекта федерального закона о техническом регламенте во втором чтении.
Федеральный орган исполнительной власти по техническому регулированию обязан опубликовать в своем печатном издании проект федерального закона о техническом регламенте в течение десяти дней с момента оплаты его опубликования. Порядок опубликования проекта федерального закона о техническом регламенте и размер платы за его опубликование устанавливаются Правительством Российской Федерации.
Проект федерального закона о техническом регламенте, подготовленный ко второму чтению, направляется Государственной Думой в Правительство Российской Федерации. На проект федерального закона о техническом регламенте Правительство Российской Федерации в течение шестидесяти дней направляет в Государственную Думу отзыв, подготовленный с учетом заключения экспертной комиссии по техническому регулированию. Проект федерального закона о техническом регламенте может быть рассмотрен Государственной Думой во втором чтении без отзыва Правительства Российской Федерации в случае, если отзыв Правительства Российской Федерации не был представлен в Государственную Думу в указанный срок.
(в ред. Федеральных законов от 01.05.2007 N 65-ФЗ, от 18.07.2009 N 189-ФЗ)
8.1. Проект постановления Правительства Российской Федерации о техническом регламенте, разработанный в установленном пунктами 2 - 6 настоящей статьи порядке и подготовленный к рассмотрению на заседании Правительства Российской Федерации, не позднее чем за тридцать дней до дня его рассмотрения направляется на экспертизу в соответствующую экспертную комиссию по техническому регулированию, которая создана и осуществляет свою деятельность в порядке, установленном пунктом 9 настоящей статьи. Проект постановления Правительства Российской Федерации о техническом регламенте рассматривается на заседании Правительства Российской Федерации с учетом заключения соответствующей экспертной комиссии по техническому регулированию.
Проект постановления Правительства Российской Федерации о техническом регламенте должен быть опубликован в печатном издании федерального органа исполнительной власти по техническому регулированию и размещен в информационной системе общего пользования в электронно-цифровой форме не позднее чем за тридцать дней до дня его рассмотрения на заседании Правительства Российской Федерации. Порядок опубликования и размещения указанного проекта постановления устанавливается Правительством Российской Федерации.
(п. 8.1 введен Федеральным законом от 01.05.2007 N 65-ФЗ)
9. Экспертиза проектов технических регламентов осуществляется экспертными комиссиями по техническому регулированию, в состав которых на паритетных началах включаются представители федеральных органов исполнительной власти, научных организаций, саморегулируемых организаций, общественных объединений предпринимателей и потребителей. Порядок создания и деятельности экспертных комиссий по техническому регулированию утверждается Правительством Российской Федерации. Федеральным органом исполнительной власти по техническому регулированию утверждается персональный состав экспертных комиссий по техническому регулированию и осуществляется обеспечение их деятельности. Заседания экспертных комиссий по техническому регулированию являются открытыми.
Заключения экспертных комиссий по техническому регулированию подлежат обязательному опубликованию в печатном издании федерального органа исполнительной власти по техническому регулированию и в информационной системе общего пользования в электронно-цифровой форме. Порядок опубликования таких заключений и размер платы за их опубликование устанавливаются Правительством Российской Федерации.
10. В случае несоответствия технического регламента интересам национальной экономики, развитию материально-технической базы и уровню научно-технического развития, а также международным нормам и правилам, введенным в действие в Российской Федерации в установленном порядке, Правительство Российской Федерации или федеральный орган исполнительной власти по техническому регулированию обязаны начать процедуру внесения изменений в технический регламент или отмены технического регламента.
(в ред. Федеральных законов от 01.05.2007 N 65-ФЗ, от 30.12.2009 N 385-ФЗ)
Внесение изменений и дополнений в технический регламент или его отмена осуществляется в порядке, предусмотренном настоящей статьей и статьей 10 настоящего Федерального закона в части разработки и принятия технических регламентов.
Статья 9.1. Порядок разработки, принятия, изменения и отмены технического регламента, принимаемого нормативным правовым актом федерального органа исполнительной власти по техническому регулированию.
1. В соответствии с поручениями Президента Российской Федерации или Правительства Российской Федерации технический регламент может быть принят нормативным правовым актом федерального органа исполнительной власти по техническому регулированию. Такой технический регламент разрабатывается в порядке, установленном пунктами 2 - 6 статьи 9 настоящего Федерального закона и настоящей статьей, и принимается в порядке, установленном для принятия нормативных правовых актов федеральных органов исполнительной власти.
(в ред. Федерального закона от 21.07.2011 N 255-ФЗ)
2. Проект технического регламента, принимаемый в форме нормативного правового акта федерального органа исполнительной власти по техническому регулированию, представляется разработчиком в федеральный орган исполнительной власти по техническому регулированию для принятия при наличии следующих документов:
обоснование необходимости принятия технического регламента с указанием требований, которые отличаются от положений соответствующих международных стандартов или обязательных требований, действующих на территории Российской Федерации в момент разработки проекта технического регламента;
финансово-экономическое обоснование принятия технического регламента;
документы, подтверждающие опубликование уведомления о разработке проекта технического регламента в соответствии с пунктом 3 статьи 9 настоящего Федерального закона;
документы, подтверждающие опубликование уведомления о завершении публичного обсуждения проекта технического регламента в соответствии с пунктом 5 статьи 9 настоящего Федерального закона;
перечень полученных в письменной форме замечаний заинтересованных лиц.
3. Представленный в федеральный орган исполнительной власти по техническому регулированию проект технического регламента с документами, указанными в пункте 2 настоящей статьи, направляется указанным органом на экспертизу в экспертную комиссию по техническому регулированию, созданную в соответствии с пунктом 9 статьи 9 настоящего Федерального закона.
4. Заключение экспертной комиссии по техническому регулированию о возможности принятия технического регламента готовится в течение тридцати дней со дня поступления проекта технического регламента с указанными в пункте 2 настоящей статьи документами в федеральный орган исполнительной власти по техническому регулированию и должно быть опубликовано в печатном издании федерального органа исполнительной власти по техническому регулированию и размещено в информационной системе общего пользования в электронно-цифровой форме.
Порядок опубликования таких заключений и размер платы за их опубликование устанавливаются Правительством Российской Федерации.
5. На основании заключения экспертной комиссии по техническому регулированию о возможности принятия технического регламента федеральный орган исполнительной власти по техническому регулированию в течение десяти дней со дня поступления такого заключения принимает решение о принятии технического регламента или об отклонении его проекта. Отклоненный проект технического регламента с заключением экспертной комиссии по техническому регулированию должен быть возвращен разработчику в течение пяти дней со дня принятия решения об отклонении проекта технического регламента.
6. Принятый технический регламент должен быть опубликован в печатном издании федерального органа исполнительной власти по техническому регулированию и размещен в информационной системе общего пользования в электронно-цифровой форме. Порядок опубликования и размещения утверждается федеральным органом исполнительной власти по техническому регулированию.
7. Федеральный орган исполнительной власти по техническому регулированию обеспечивает в информационной системе общего пользования в электронно-цифровой форме доступ на безвозмездной основе к принятым техническим регламентам.
8. Принятые нормативным правовым актом федерального органа исполнительной власти по техническому регулированию технические регламенты подлежат государственной регистрации в установленном порядке.
9. Внесение изменений в технический регламент или его отмена осуществляется в порядке, предусмотренном настоящей статьей и статьей 10 настоящего Федерального закона в части разработки и принятия технических регламентов.
Статья 10. Особый порядок разработки и принятия технических регламентов.
1. В исключительных случаях при возникновении обстоятельств, приводящих к непосредственной угрозе жизни или здоровью граждан, окружающей среде, жизни или здоровью животных и растений, и в случаях, если для обеспечения безопасности продукции или связанных с требованиями к ней процессов проектирования (включая изыскания), производства, строительства, монтажа, наладки, эксплуатации, хранения, перевозки, реализации и утилизации необходимо незамедлительное принятие соответствующего нормативного правового акта о техническом регламенте, Президент Российской Федерации вправе издать технический регламент без его публичного обсуждения.
(в ред. Федерального закона от 01.05.2007 N 65-ФЗ)
4. Со дня вступления в силу федерального закона о техническом регламенте соответствующий технический регламент, изданный указом Президента Российской Федерации, постановлением Правительства Российской Федерации или нормативным правовым актом федерального органа исполнительной власти по техническому регулированию, утрачивает силу.
(п. 4 в ред. Федерального закона от 30.12.2009 N 385-ФЗ).
Глава 3. СТАНДАРТИЗАЦИЯ
Статья 11. Цели стандартизации
Целями стандартизации являются:
повышение уровня безопасности жизни и здоровья граждан, имущества физических и юридических лиц, государственного и муниципального имущества, объектов с учетом риска возникновения чрезвычайных ситуаций природного и техногенного характера, повышение уровня экологической безопасности, безопасности жизни и здоровья животных и растений;
обеспечение конкурентоспособности и качества продукции (работ, услуг), единства измерений, рационального использования ресурсов, взаимозаменяемости технических средств (машин и оборудования, их составных частей, комплектующих изделий и материалов), технической и информационной совместимости, сопоставимости результатов исследований (испытаний) и измерений, технических и экономико-статистических данных, проведения анализа характеристик продукции (работ, услуг), планирования и осуществления закупок товаров, работ, услуг для обеспечения государственных и муниципальных нужд, добровольного подтверждения соответствия продукции (работ, услуг);
содействие соблюдению требований технических регламентов;
создание систем классификации и кодирования технико-экономической и социальной информации, систем каталогизации продукции (работ, услуг), систем обеспечения качества продукции (работ, услуг), систем поиска и передачи данных, содействие проведению работ по унификации.
Статья 12. Принципы стандартизации
Стандартизация осуществляется в соответствии с принципами:
добровольного применения документов в области стандартизации;
максимального учета при разработке стандартов законных интересов заинтересованных лиц;
применения международного стандарта как основы разработки национального стандарта, за исключением случаев, если такое применение признано невозможным вследствие несоответствия требований международных стандартов климатическим и географическим особенностям Российской Федерации, техническим и (или) технологическим особенностям или по иным основаниям либо Российская Федерация в соответствии с установленными процедурами выступала против принятия международного стандарта или отдельного его положения;
недопустимости создания препятствий производству и обращению продукции, выполнению работ и оказанию услуг в большей степени, чем это минимально необходимо для выполнения целей, указанных в статье 11 настоящего Федерального закона;
недопустимости установления таких стандартов, которые противоречат техническим регламентам;
обеспечения условий для единообразного применения стандартов.
Статья 13. Документы в области стандартизации
К документам в области стандартизации, используемым на территории Российской Федерации, относятся:
национальные стандарты;
правила стандартизации, нормы и рекомендации в области стандартизации;
применяемые в установленном порядке классификации, общероссийские классификаторы технико-экономической и социальной информации;
стандарты организаций;
своды правил;
международные стандарты, региональные стандарты, региональные своды правил, стандарты иностранных государств и своды правил иностранных государств, зарегистрированные в Федеральном информационном фонде технических регламентов и стандартов;
надлежащим образом заверенные переводы на русский язык международных стандартов, региональных стандартов, региональных сводов правил, стандартов иностранных государств и сводов правил иностранных государств, принятые на учет национальным органом Российской Федерации по стандартизации;
предварительные национальные стандарты.
Глава 4. ПОДТВЕРЖДЕНИЕ СООТВЕТСТВИЯ
Статья 18. Цели подтверждения соответствия
Подтверждение соответствия осуществляется в целях:
удостоверения соответствия продукции, процессов проектирования (включая изыскания), производства, строительства, монтажа, наладки, эксплуатации, хранения, перевозки, реализации и утилизации, работ, услуг или иных объектов техническим регламентам, стандартам, сводам правил, условиям договоров;
содействия приобретателям, в том числе потребителям, в компетентном выборе продукции, работ, услуг;
повышения конкурентоспособности продукции, работ, услуг на российском и международном рынках;
создания условий для обеспечения свободного перемещения товаров по территории Российской Федерации, а также для осуществления международного экономического, научно-технического сотрудничества и международной торговли.
Статья 19. Принципы подтверждения соответствия
1. Подтверждение соответствия осуществляется на основе принципов:
доступности информации о порядке осуществления подтверждения соответствия заинтересованным лицам;
недопустимости применения обязательного подтверждения соответствия к объектам, в отношении которых не установлены требования технических регламентов;
установления перечня форм и схем обязательного подтверждения соответствия в отношении определенных видов продукции в соответствующем техническом регламенте;
уменьшения сроков осуществления обязательного подтверждения соответствия и затрат заявителя;
недопустимости принуждения к осуществлению добровольного подтверждения соответствия, в том числе в определенной системе добровольной сертификации;
защиты имущественных интересов заявителей, соблюдения коммерческой тайны в отношении сведений, полученных при осуществлении подтверждения соответствия;
недопустимости подмены обязательного подтверждения соответствия добровольной сертификацией.
2. Подтверждение соответствия разрабатывается и применяется равным образом и в равной мере независимо от страны и (или) места происхождения продукции, осуществления процессов проектирования (включая изыскания), производства, строительства, монтажа, наладки, эксплуатации, хранения, перевозки, реализации и утилизации, выполнения работ и оказания услуг, видов или особенностей сделок и (или) лиц, которые являются изготовителями, исполнителями, продавцами, приобретателями.
Статья 20. Формы подтверждения соответствия
1. Подтверждение соответствия на территории Российской Федерации может носить добровольный или обязательный характер.
2. Добровольное подтверждение соответствия осуществляется в форме добровольной сертификации.
3. Обязательное подтверждение соответствия осуществляется в формах:
принятия декларации о соответствии (далее - декларирование соответствия);
обязательной сертификации.
4. Порядок применения форм обязательного подтверждения соответствия устанавливается настоящим Федеральным законом.
Статья 21. Добровольное подтверждение соответствия
1. Добровольное подтверждение соответствия осуществляется по инициативе заявителя на условиях договора между заявителем и органом по сертификации. Добровольное подтверждение соответствия может осуществляться для установления соответствия национальным стандартам, предварительным национальным стандартам, стандартам организаций, сводам правил, системам добровольной сертификации, условиям договоров.
Объектами добровольного подтверждения соответствия являются продукция, процессы производства, эксплуатации, хранения, перевозки, реализации и утилизации, работы и услуги, а также иные объекты, в отношении которых стандартами, системами добровольной сертификации и договорами устанавливаются требования.
Орган по сертификации:
осуществляет подтверждение соответствия объектов добровольного подтверждения соответствия;
выдает сертификаты соответствия на объекты, прошедшие добровольную сертификацию;
предоставляет заявителям право на применение знака соответствия, если применение знака соответствия предусмотрено соответствующей системой добровольной сертификации;
приостанавливает или прекращает действие выданных им сертификатов соответствия.
2. Система добровольной сертификации может быть создана юридическим лицом и (или) индивидуальным предпринимателем или несколькими юридическими лицами и (или) индивидуальными предпринимателями.
Лицо или лица, создавшие систему добровольной сертификации, устанавливают перечень объектов, подлежащих сертификации, и их характеристик, на соответствие которым осуществляется добровольная сертификация, правила выполнения предусмотренных данной системой добровольной сертификации работ и порядок их оплаты, определяют участников данной системы добровольной сертификации. Системой добровольной сертификации может предусматриваться применение знака соответствия.
3. Система добровольной сертификации может быть зарегистрирована федеральным органом исполнительной власти по техническому регулированию.
Для регистрации системы добровольной сертификации в федеральный орган исполнительной власти по техническому регулированию представляются:
свидетельство о государственной регистрации юридического лица и (или) индивидуального предпринимателя. В случае, если указанный документ не представлен лицом или лицами, создавшими систему добровольной сертификации, по собственной инициативе, сведения, содержащиеся в нем, представляются уполномоченным федеральным органом исполнительной власти по межведомственному запросу федерального органа исполнительной власти по техническому регулированию;
правила функционирования системы добровольной сертификации, которыми предусмотрены положения пункта 2 настоящей статьи;
изображение знака соответствия, применяемое в данной системе добровольной сертификации, если применение знака соответствия предусмотрено, и порядок применения знака соответствия;
документ об оплате регистрации системы добровольной сертификации.
Регистрация системы добровольной сертификации осуществляется в течение пяти дней с момента представления документов, предусмотренных настоящим пунктом для регистрации системы добровольной сертификации, в федеральный орган исполнительной власти по техническому регулированию. Порядок регистрации системы добровольной сертификации и размер платы за регистрацию устанавливаются Правительством Российской Федерации. Плата за регистрацию системы добровольной сертификации подлежит зачислению в федеральный бюджет.
4. Отказ в регистрации системы добровольной сертификации допускается только в случае непредставления документов, предусмотренных абзацами четвертым, пятым и шестым пункта 3 настоящей статьи, отсутствия сведений о государственной регистрации юридического лица и (или) индивидуального предпринимателя или совпадения наименования системы и (или) изображения знака соответствия с наименованием системы и (или) изображением знака соответствия зарегистрированной ранее системы добровольной сертификации. Уведомление об отказе в регистрации системы добровольной сертификации направляется заявителю в течение трех дней со дня принятия решения об отказе в регистрации этой системы с указанием оснований для отказа.
Отказ в регистрации системы добровольной сертификации может быть обжалован в судебном порядке.
5. Федеральный орган исполнительной власти по техническому регулированию ведет единый реестр зарегистрированных систем добровольной сертификации, содержащий сведения о юридических лицах и (или) об индивидуальных предпринимателях, создавших системы добровольной сертификации, о правилах функционирования систем добровольной сертификации, которыми предусмотрены положения пункта 2 настоящей статьи, знаках соответствия и порядке их применения. Федеральный орган исполнительной власти по техническому регулированию должен обеспечить доступность сведений, содержащихся в едином реестре зарегистрированных систем добровольной сертификации, заинтересованным лицам.
Порядок ведения единого реестра зарегистрированных систем добровольной сертификации и порядок предоставления сведений, содержащихся в этом реестре, устанавливаются федеральным органом исполнительной власти по техническому регулированию.
Статья 22. Знаки соответствия
1. Объекты сертификации, сертифицированные в системе добровольной сертификации, могут маркироваться знаком соответствия системы добровольной сертификации. Порядок применения такого знака соответствия устанавливается правилами соответствующей системы добровольной сертификации.
2. Применение знака соответствия национальному стандарту осуществляется заявителем на добровольной основе любым удобным для заявителя способом в порядке, установленном национальным органом по стандартизации.
3. Объекты, соответствие которых не подтверждено в порядке, установленном настоящим Федеральным законом, не могут быть маркированы знаком соответствия.
Статья 23. Обязательное подтверждение соответствия
1. Обязательное подтверждение соответствия проводится только в случаях, установленных соответствующим техническим регламентом, и исключительно на соответствие требованиям технического регламента.
Объектом обязательного подтверждения соответствия может быть только продукция, выпускаемая в обращение на территории Российской Федерации.
2. Форма и схемы обязательного подтверждения соответствия могут устанавливаться только техническим регламентом с учетом степени риска недостижения целей технических регламентов.
3. Декларация о соответствии и сертификат соответствия имеют равную юридическую силу и действуют на всей территории Российской Федерации в отношении каждой единицы продукции, выпускаемой в обращение на территории Российской Федерации во время действия декларации о соответствии или сертификата соответствия, в течение срока годности или срока службы продукции, установленных в соответствии с законодательством Российской Федерации.
4. Работы по обязательному подтверждению соответствия подлежат оплате на основании договора с заявителем. Стоимость работ по обязательному подтверждению соответствия продукции определяется независимо от страны и (или) места ее происхождения, а также лиц, которые являются заявителями.
Статья 24. Декларирование соответствия
1. Декларирование соответствия осуществляется по одной из следующих схем:
принятие декларации о соответствии на основании собственных доказательств;
принятие декларации о соответствии на основании собственных доказательств, доказательств, полученных с участием органа по сертификации и (или) аккредитованной испытательной лаборатории (центра) (далее - третья сторона).
При декларировании соответствия заявителем может быть зарегистрированные в соответствии с законодательством Российской Федерации на ее территории юридическое лицо или физическое лицо в качестве индивидуального предпринимателя, либо являющиеся изготовителем или продавцом, либо выполняющие функции иностранного изготовителя на основании договора с ним в части обеспечения соответствия поставляемой продукции требованиям технических регламентов и в части ответственности за несоответствие поставляемой продукции требованиям технических регламентов (лицо, выполняющее функции иностранного изготовителя).
Круг заявителей устанавливается соответствующим техническим регламентом.
Схема декларирования соответствия с участием третьей стороны устанавливается в техническом регламенте в случае, если отсутствие третьей стороны приводит к недостижению целей подтверждения соответствия.
2. При декларировании соответствия заявитель на основании собственных доказательств самостоятельно формирует доказательственные материалы в целях подтверждения соответствия продукции требованиям технического регламента. В качестве доказательственных материалов используются техническая документация, результаты собственных исследований (испытаний) и измерений и (или) другие документы, послужившие основанием для подтверждения соответствия продукции требованиям технического регламента.
Техническая документация должна содержать:
основные параметры и характеристики продукции, а также ее описание в целях оценки соответствия продукции требованиям технического регламента;
описание мер по обеспечению безопасности продукции на одной или нескольких стадиях проектирования (включая изыскания), производства, строительства, монтажа, наладки, эксплуатации, хранения, перевозки, реализации и утилизации;
список документов в области стандартизации, применяемых полностью или частично и включенных в перечень документов в области стандартизации, в результате применения которых на добровольной основе обеспечивается соблюдение требований технического регламента, и, если не применялись указанные документы в области стандартизации, описание решений, выбранных для реализации требований технического регламента. В случае, если документы в области стандартизации, включенные в перечень документов в области стандартизации, в результате применения которых на добровольной основе обеспечивается соблюдение требований технического регламента, применялись частично, в технической документации указываются применяемые разделы указанных документов.
Техническая документация также может содержать общее описание продукции, конструкторскую и технологическую документацию на продукцию, схемы компонентов, узлов, цепей, описания и пояснения, необходимые для понимания указанных схем, а также результаты выполненных проектных расчетов, проведенного контроля, иные документы, послужившие мотивированным основанием для подтверждения соответствия продукции требованиям технического регламента.
Техническая документация, используемая в качестве доказательственного материала, также может содержать анализ риска применения (использования) продукции. Состав доказательственных материалов определяется соответствующим техническим регламентом, состав указанной технической документации может уточняться соответствующим техническим регламентом.
3. При декларировании соответствия на основании собственных доказательств и полученных с участием третьей стороны доказательств заявитель по своему выбору в дополнение к собственным доказательствам, сформированным в порядке, предусмотренном пунктом 2 настоящей статьи:
включает в доказательственные материалы протоколы исследований (испытаний) и измерений, проведенных в аккредитованной испытательной лаборатории (центре);
предоставляет сертификат системы менеджмента качества, в отношении которого предусматривается контроль (надзор) органа по сертификации, выдавшего данный сертификат, за объектом сертификации.
4.1. При декларировании соответствия заявитель, не применяющий документов в области стандартизации, включенных в перечень документов в области стандартизации, в результате применения которых на добровольной основе обеспечивается соблюдение требований технического регламента, может обратиться в орган по сертификации за заключением о соответствии его продукции требованиям технического регламента и на основании указанного заключения органа по сертификации, подготовленного по результатам проведенных исследований (испытаний), измерений типового образца выпускаемой продукции, технической документации на данную продукцию, принять декларацию о соответствии в порядке, установленном пунктом 2 настоящей статьи или соответствующим техническим регламентом.
5. Декларация о соответствии оформляется на русском языке и должна содержать:
наименование и местонахождение заявителя;
наименование и местонахождение изготовителя;
информацию об объекте подтверждения соответствия, позволяющую идентифицировать этот объект;
наименование технического регламента, на соответствие требованиям которого подтверждается продукция;
указание на схему декларирования соответствия;
заявление заявителя о безопасности продукции при ее использовании в соответствии с целевым назначением и принятии заявителем мер по обеспечению соответствия продукции требованиям технических регламентов;
сведения о проведенных исследованиях (испытаниях) и измерениях, сертификате системы менеджмента качества, а также документах, послуживших основанием для подтверждения соответствия продукции требованиям технических регламентов;
срок действия декларации о соответствии;
иные предусмотренные соответствующими техническими регламентами сведения.
Срок действия декларации о соответствии определяется техническим регламентом.
Форма декларации о соответствии утверждается федеральным органом исполнительной власти по техническому регулированию.
6. Оформленная заявителем в соответствии с пунктом 5 настоящей статьи декларация о соответствии подлежит регистрации в электронной форме в едином реестре деклараций о соответствии в уведомительном порядке в течение трех дней со дня ее принятия.
Ведение единого реестра деклараций о соответствии осуществляет федеральный орган исполнительной власти, уполномоченный Правительством Российской Федерации.
Порядок формирования и ведения единого реестра деклараций о соответствии и порядок регистрации деклараций о соответствии устанавливаются федеральным органом исполнительной власти, уполномоченным Правительством Российской Федерации.
7. Декларация о соответствии и доказательственные материалы хранятся у заявителя в течение десяти лет со дня окончания срока действия такой декларации в случае, если иной срок их хранения не установлен техническим регламентом. Заявитель обязан представить декларацию о соответствии и доказательственные материалы по требованию федерального органа исполнительной власти, уполномоченного на осуществление государственного контроля (надзора) за соблюдением требований технических регламентов.
Статья 25. Обязательная сертификация
1. Обязательная сертификация осуществляется органом по сертификации на основании договора с заявителем. Схемы сертификации, применяемые для сертификации определенных видов продукции, устанавливаются соответствующим техническим регламентом. Круг заявителей устанавливается соответствующим техническим регламентом.
2. Соответствие продукции требованиям технических регламентов подтверждается сертификатом соответствия, выдаваемым заявителю органом по сертификации.
Сертификат соответствия включает в себя:
наименование и местонахождение заявителя;
наименование и местонахождение изготовителя продукции, прошедшей сертификацию;
наименование и местонахождение органа по сертификации, выдавшего сертификат соответствия;
информацию об объекте сертификации, позволяющую идентифицировать этот объект;
наименование технического регламента, на соответствие требованиям которого проводилась сертификация;
информацию о проведенных исследованиях (испытаниях) и измерениях;
информацию о документах, представленных заявителем в орган по сертификации в качестве доказательств соответствия продукции требованиям технических регламентов;
срок действия сертификата соответствия;
информацию об использовании или о неиспользовании заявителем национальных стандартов, включенных в перечень документов в области стандартизации, в результате применения которых на добровольной основе обеспечивается соблюдение требований технического регламента.
Сертификат соответствия выдается на серийно выпускаемую продукцию, на отдельно поставляемую партию продукции или на единичный экземпляр продукции.
Срок действия сертификата соответствия определяется соответствующим техническим регламентом.
Форма сертификата соответствия утверждается федеральным органом исполнительной власти по техническому регулированию.
3. В случае, если в отношении впервые выпускаемой в обращение продукции отсутствуют или не могут быть применены документы в области стандартизации, в результате применения которых на добровольной основе обеспечивается соблюдение требований технического регламента, и такая продукция относится к виду, типу продукции, подлежащей обязательной сертификации, изготовитель (лицо, выполняющее функции иностранного изготовителя) вправе осуществить декларирование ее соответствия на основании собственных доказательств. При декларировании соответствия такой продукции изготовитель (лицо, выполняющее функции иностранного изготовителя) указывает в декларации о соответствии, в сопроводительной документации и при маркировке такой продукции сведения о том, что обязательная сертификация такой продукции не осуществлялась.
В случае, если в отношении впервые выпускаемой в обращение продукции отсутствуют или не могут быть применены документы в области стандартизации, в результате применения которых на добровольной основе обеспечивается соблюдение требований технического регламента, и такая продукция относится к виду, типу продукции, в отношении которой предусмотрено декларирование соответствия на основании доказательств, полученных с участием третьей стороны, изготовитель (лицо, выполняющее функции иностранного изготовителя) вправе осуществить декларирование ее соответствия на основании собственных доказательств. При декларировании соответствия такой продукции изготовитель (лицо, выполняющее функции иностранного изготовителя) указывает в декларации о соответствии, в сопроводительной документации и при маркировке такой продукции сведения об отсутствии у него доказательств, полученных с участием третьей стороны.
Особенности маркировки впервые выпускаемой в обращение продукции, в том числе знаком обращения на рынке, порядок информирования приобретателя, в том числе потребителя, о возможном вреде такой продукции и о факторах, от которых он зависит, определяются Правительством Российской Федерации.
Статья 26. Организация обязательной сертификации
1. Обязательная сертификация осуществляется органом по сертификации, аккредитованным в порядке, установленном Правительством Российской Федерации.
2. Орган по сертификации:
привлекает на договорной основе для проведения исследований (испытаний) и измерений аккредитованные испытательные лаборатории (центры);
осуществляет контроль за объектами сертификации, если такой контроль предусмотрен соответствующей схемой обязательной сертификации и договором;
ведет реестр выданных им сертификатов соответствия;
информирует соответствующие органы государственного контроля (надзора) за соблюдением требований технических регламентов о продукции, поступившей на сертификацию, но не прошедшей ее;
выдает сертификаты соответствия, приостанавливает или прекращает действие выданных им сертификатов соответствия и информирует об этом федеральный орган исполнительной власти, организующий формирование и ведение единого реестра сертификатов соответствия, и органы государственного контроля (надзора) за соблюдением требований технических регламентов;
обеспечивает предоставление заявителям информации о порядке проведения обязательной сертификации;
определяет стоимость работ по сертификации, выполняемых в соответствии с договором с заявителем;
в порядке, установленном соответствующим техническим регламентом, принимает решение о продлении срока действия сертификата соответствия, в том числе по результатам проведенного контроля за сертифицированными объектами;
осуществляет отбор образцов для целей сертификации и представляет их для проведения исследований (испытаний) и измерений в аккредитованные испытательные лаборатории (центры) или поручает осуществить такой отбор аккредитованным испытательным лабораториям (центрам);
подготавливает заключение, на основании которого заявитель вправе принять декларацию о соответствии по результатам проведенных исследований (испытаний), измерений типовых образцов выпускаемой в обращение продукции и технической документации на данную продукцию.
3. Порядок формирования и ведения единого реестра сертификатов соответствия, порядок предоставления содержащихся в указанном реестре сведений и оплаты за их предоставление, а также федеральный орган исполнительной власти, организующий формирование и ведение указанного реестра, определяется Правительством Российской Федерации.
4. Исследования (испытания) и измерения продукции при осуществлении обязательной сертификации проводятся аккредитованными испытательными лабораториями (центрами).
Аккредитованные испытательные лаборатории (центры) проводят исследования (испытания) и измерения продукции в пределах своей области аккредитации на условиях договоров с органами по сертификации. Органы по сертификации не вправе предоставлять аккредитованным испытательным лабораториям (центрам) сведения о заявителе.
Аккредитованная испытательная лаборатория (центр) оформляет результаты исследований (испытаний) и измерений соответствующими протоколами, на основании которых орган по сертификации принимает решение о выдаче или об отказе в выдаче сертификата соответствия. Аккредитованная испытательная лаборатория (центр) обязана обеспечить достоверность результатов исследований (испытаний) и измерений.
ФЕДЕРАЛЬНЫЙ ЗАКОН
ОБ ОБРАЩЕНИИ ЛЕКАРСТВЕННЫХ СРЕДСТВ
(извлечение)
(в ред. Федеральных законов от 02.07.2013 N 185-ФЗ,
от 25.11.2013 N 317-ФЗ, от 12.03.2014 N 33-ФЗ)
Глава 1. ОБЩИЕ ПОЛОЖЕНИЯ
Статья 1. Предмет регулирования настоящего Федерального закона
1. Настоящий Федеральный закон регулирует отношения, возникающие в связи с обращением - разработкой, доклиническими исследованиями, клиническими исследованиями, экспертизой, государственной регистрацией, со стандартизацией и с контролем качества, производством, изготовлением, хранением, перевозкой, ввозом в Российскую Федерацию, вывозом из Российской Федерации, рекламой, отпуском, реализацией, передачей, применением, уничтожением лекарственных средств.
2. Настоящий Федеральный закон устанавливает приоритет государственного регулирования безопасности, качества и эффективности лекарственных средств при их обращении.
Статья 2. Сфера применения настоящего Федерального закона
Настоящий Федеральный закон применяется к отношениям, возникающим при обращении лекарственных средств на территории Российской Федерации.
Статья 3. Законодательство об обращении лекарственных средств
1. Законодательство об обращении лекарственных средств состоит из настоящего Федерального закона, других федеральных законов и иных нормативных правовых актов Российской Федерации.
2. Действие настоящего Федерального закона распространяется на обращение наркотических лекарственных средств и психотропных лекарственных средств с учетом особенностей, установленных законодательством Российской Федерации о наркотических средствах, психотропных веществах и об их прекурсорах.
3. Действие настоящего Федерального закона распространяется на обращение радиофармацевтических лекарственных средств с учетом особенностей, установленных законодательством Российской Федерации в области обеспечения радиационной безопасности.
4. Если международным договором Российской Федерации установлены иные правила, чем те, которые предусмотрены настоящим Федеральным законом, применяются правила международного договора.
5. В Российской Федерации в соответствии с международными договорами Российской Федерации и (или) на основе принципа взаимности признаются результаты клинических исследований лекарственных препаратов для медицинского применения, проведенных за пределами территории Российской Федерации.
Статья 4. Основные понятия, используемые в настоящем Федеральном законе
Для целей настоящего Федерального закона используются следующие основные понятия:
1) лекарственные средства - вещества или их комбинации, вступающие в контакт с организмом человека или животного, проникающие в органы, ткани организма человека или животного, применяемые для профилактики, диагностики (за исключением веществ или их комбинаций, не контактирующих с организмом человека или животного), лечения заболевания, реабилитации, для сохранения, предотвращения или прерывания беременности и полученные из крови, плазмы крови, из органов, тканей организма человека или животного, растений, минералов методами синтеза или с применением биологических технологий. К лекарственным средствам относятся фармацевтические субстанции и лекарственные препараты;
2) фармацевтические субстанции - лекарственные средства в виде действующих веществ биологического, биотехнологического, минерального или химического происхождения, обладающие фармакологической активностью, предназначенные для производства, изготовления лекарственных препаратов и определяющие их эффективность;
3) вспомогательные вещества - вещества неорганического или органического происхождения, используемые в процессе производства, изготовления лекарственных препаратов для придания им необходимых физико-химических свойств;
4) лекарственные препараты - лекарственные средства в виде лекарственных форм, применяемые для профилактики, диагностики, лечения заболевания, реабилитации, для сохранения, предотвращения или прерывания беременности;
5) лекарственная форма - состояние лекарственного препарата, соответствующее способам его введения и применения и обеспечивающее достижение необходимого лечебного эффекта;
6) перечень жизненно необходимых и важнейших лекарственных препаратов - ежегодно утверждаемый Правительством Российской Федерации перечень лекарственных препаратов для медицинского применения, обеспечивающих приоритетные потребности здравоохранения в целях профилактики и лечения заболеваний, в том числе преобладающих в структуре заболеваемости в Российской Федерации;
7) иммунобиологические лекарственные препараты - лекарственные препараты биологического происхождения, предназначенные для иммунологических диагностики, профилактики и лечения заболеваний;
8) наркотические лекарственные средства - лекарственные препараты и фармацевтические субстанции, содержащие наркотические средства и включенные в Перечень наркотических средств, психотропных веществ и их прекурсоров, подлежащих контролю в Российской Федерации, в соответствии с законодательством Российской Федерации, международными договорами Российской Федерации, в том числе Единой конвенцией о наркотических средствах 1961 года;
9) психотропные лекарственные средства - лекарственные препараты и фармацевтические субстанции, содержащие психотропные вещества и включенные в Перечень наркотических средств, психотропных веществ и их прекурсоров, подлежащих контролю в Российской Федерации, в соответствии с законодательством Российской Федерации, международными договорами Российской Федерации, в том числе Конвенцией о психотропных веществах 1971 года;
10) радиофармацевтические лекарственные средства - лекарственные средства, которые содержат в готовой для использования форме один радионуклид или несколько радионуклидов (радиоактивных изотопов);
11) оригинальное лекарственное средство - лекарственное средство, содержащее впервые полученную фармацевтическую субстанцию или новую комбинацию фармацевтических субстанций, эффективность и безопасность которых подтверждены результатами доклинических исследований лекарственных средств и клинических исследований лекарственных препаратов;
12) воспроизведенное лекарственное средство - лекарственное средство, содержащее такую же фармацевтическую субстанцию или комбинацию таких же фармацевтических субстанций в такой же лекарственной форме, что и оригинальное лекарственное средство, и поступившее в обращение после поступления в обращение оригинального лекарственного средства;
13) лекарственное растительное сырье - свежие или высушенные растения либо их части, используемые для производства лекарственных средств организациями - производителями лекарственных средств или изготовления лекарственных препаратов аптечными организациями, ветеринарными аптечными организациями, индивидуальными предпринимателями, имеющими лицензию на фармацевтическую деятельность;
14) лекарственный растительный препарат - лекарственный препарат, произведенный или изготовленный из одного вида лекарственного растительного сырья или нескольких видов такого сырья и реализуемый в расфасованном виде во вторичной (потребительской) упаковке;
15) гомеопатическое лекарственное средство - лекарственное средство, произведенное или изготовленное по специальной технологии;
16) международное непатентованное наименование лекарственного средства - наименование фармацевтической субстанции, рекомендованное Всемирной организацией здравоохранения;
17) торговое наименование лекарственного средства - наименование лекарственного средства, присвоенное его разработчиком;
18) общая фармакопейная статья - документ, утвержденный уполномоченным федеральным органом исполнительной власти и содержащий перечень показателей качества и (или) методов контроля качества конкретной лекарственной формы, лекарственного растительного сырья, описания биологических, биохимических, микробиологических, физико-химических, физических, химических и других методов анализа лекарственного средства для медицинского применения, а также требования к используемым в целях проведения данного анализа реактивам, титрованным растворам, индикаторам;
19) фармакопейная статья - документ, утвержденный уполномоченным федеральным органом исполнительной власти и содержащий перечень показателей качества и методов контроля качества лекарственного средства для медицинского применения;
20) нормативная документация - документ, содержащий перечень определяемых по результатам соответствующих экспертиз показателей качества лекарственного средства для медицинского применения, методов контроля его качества и установленный его производителем;
21) нормативный документ - документ, содержащий перечень определяемых по результатам соответствующих экспертиз показателей качества и (или) методов контроля качества лекарственной формы, описания биологических, биохимических, микробиологических, физико-химических, физических, химических и других методов анализа лекарственных средств для ветеринарного применения, требования к используемым в целях проведения данного анализа реактивам, титрованным растворам, индикаторам и установленный его производителем;
22) качество лекарственного средства - соответствие лекарственного средства требованиям фармакопейной статьи либо в случае ее отсутствия нормативной документации или нормативного документа;
23) безопасность лекарственного средства - характеристика лекарственного средства, основанная на сравнительном анализе его эффективности и риска причинения вреда здоровью;
24) эффективность лекарственного препарата - характеристика степени положительного влияния лекарственного препарата на течение, продолжительность заболевания или его предотвращение, реабилитацию, на сохранение, предотвращение или прерывание беременности;
25) серия лекарственного средства - количество лекарственного средства, произведенное в результате одного технологического цикла его производителем;
26) регистрационное удостоверение лекарственного препарата - документ, подтверждающий факт государственной регистрации лекарственного препарата;
27) регистрационный номер - кодовое обозначение, присвоенное лекарственному препарату при его государственной регистрации;
28) обращение лекарственных средств - разработка, доклинические исследования, клинические исследования, экспертиза, государственная регистрация, стандартизация и контроль качества, производство, изготовление, хранение, перевозка, ввоз в Российскую Федерацию, вывоз из Российской Федерации, реклама, отпуск, реализация, передача, применение, уничтожение лекарственных средств;
29) субъекты обращения лекарственных средств - физические лица, в том числе индивидуальные предприниматели, и юридические лица, осуществляющие деятельность при обращении лекарственных средств;
30) разработчик лекарственного средства - организация, обладающая правами на результаты доклинических исследований лекарственного средства, клинических исследований лекарственного препарата, а также на технологию производства лекарственного средства;
31) производство лекарственных средств - деятельность по производству лекарственных средств организациями - производителями лекарственных средств на одной стадии, нескольких или всех стадиях технологического процесса, а также по хранению и реализации произведенных лекарственных средств;
32) производитель лекарственных средств - организация, осуществляющая производство лекарственных средств в соответствии с требованиями настоящего Федерального закона;
33) фармацевтическая деятельность - деятельность, включающая в себя оптовую торговлю лекарственными средствами, их хранение, перевозку и (или) розничную торговлю лекарственными препаратами, их отпуск, хранение, перевозку, изготовление лекарственных препаратов;
34) организация оптовой торговли лекарственными средствами - организация, осуществляющая оптовую торговлю лекарственными средствами, их хранение, перевозку в соответствии с требованиями настоящего Федерального закона;
35) аптечная организация - организация, структурное подразделение медицинской организации, осуществляющие розничную торговлю лекарственными препаратами, хранение, перевозку, изготовление и отпуск лекарственных препаратов для медицинского применения в соответствии с требованиями настоящего Федерального закона;
36) ветеринарная аптечная организация - организация, структурное подразделение ветеринарной организации, осуществляющие розничную торговлю лекарственными препаратами, хранение, изготовление и отпуск лекарственных препаратов для ветеринарного применения в соответствии с требованиями настоящего Федерального закона;
37) фальсифицированное лекарственное средство - лекарственное средство, сопровождаемое ложной информацией о его составе и (или) производителе;
38) недоброкачественное лекарственное средство - лекарственное средство, не соответствующее требованиям фармакопейной статьи либо в случае ее отсутствия требованиям нормативной документации или нормативного документа;
39) контрафактное лекарственное средство - лекарственное средство, находящееся в обороте с нарушением гражданского законодательства;
40) доклиническое исследование лекарственного средства - биологические, микробиологические, иммунологические, токсикологические, фармакологические, физические, химические и другие исследования лекарственного средства путем применения научных методов оценок в целях получения доказательств безопасности, качества и эффективности лекарственного средства;
41) клиническое исследование лекарственного препарата - изучение диагностических, лечебных, профилактических, фармакологических свойств лекарственного препарата в процессе его применения у человека, животного, в том числе процессов всасывания, распределения, изменения и выведения, путем применения научных методов оценок в целях получения доказательств безопасности, качества и эффективности лекарственного препарата, данных о нежелательных реакциях организма человека, животного на применение лекарственного препарата и об эффекте его взаимодействия с другими лекарственными препаратами и (или) пищевыми продуктами, кормами;
42) многоцентровое клиническое исследование лекарственного препарата для медицинского применения - клиническое исследование лекарственного препарата для медицинского применения, проводимое разработчиком лекарственного препарата в двух и более медицинских организациях по единому протоколу клинического исследования лекарственного препарата;
43) международное многоцентровое клиническое исследование лекарственного препарата для медицинского применения - клиническое исследование лекарственного препарата для медицинского применения, проводимое разработчиком лекарственного препарата в различных странах по единому протоколу клинического исследования лекарственного препарата;
44) пострегистрационное клиническое исследование лекарственного препарата для медицинского применения - клиническое исследование лекарственного препарата для медицинского применения, проводимое производителем лекарственного препарата, гражданский оборот которого осуществляется после государственной регистрации, в целях дополнительного сбора данных о его безопасности и эффективности, расширения показаний к применению данного лекарственного препарата, а также выявления нежелательных реакций пациентов на его действие;
45) исследование биоэквивалентности лекарственного препарата - вид клинического исследования лекарственного препарата, проведение которого осуществляется для определения скорости всасывания и выведения фармацевтической субстанции, количества фармацевтической субстанции, достигающего системного кровотока, и результаты которого позволяют сделать вывод о биоэквивалентности воспроизведенного лекарственного препарата в определенных лекарственной форме и дозировке соответствующему оригинальному лекарственному препарату;
46) исследование терапевтической эквивалентности лекарственных препаратов - вид клинического исследования лекарственных препаратов, проведение которого осуществляется для выявления одинаковых свойств лекарственных препаратов определенной лекарственной формы, а также наличия одинаковых показателей безопасности и эффективности лекарственных препаратов, одинаковых клинических эффектов при их применении;
47) протокол клинического исследования лекарственного препарата - документ, в котором определяются цели, формы организации и методология проведения клинического исследования, статистические методы обработки результатов такого исследования и меры по обеспечению безопасности физических лиц, участвующих в клиническом исследовании лекарственного препарата;
48) брошюра исследователя - сводное изложение результатов доклинического исследования лекарственного средства и клинического исследования лекарственного препарата для медицинского применения;
49) информационный листок пациента - документ, в котором содержатся в доступной форме сведения, касающиеся проводимого клинического исследования лекарственного препарата, и в письменной форме добровольное согласие пациента на участие в клиническом исследовании лекарственного препарата после ознакомления с особенностями клинического исследования, имеющими значение для выражения такого согласия;
50) побочное действие - реакция организма, возникшая в связи с применением лекарственного препарата в дозах, рекомендуемых в инструкции по его применению, для профилактики, диагностики, лечения заболевания или для реабилитации;
51) серьезная нежелательная реакция - нежелательная реакция организма, связанная с применением лекарственного препарата, приведшая к смерти, врожденным аномалиям или порокам развития либо представляющая собой угрозу жизни, требующая госпитализации или приведшая к стойкой утрате трудоспособности и (или) инвалидности;
52) непредвиденная нежелательная реакция - нежелательная реакция организма (в том числе связанная с применением лекарственного препарата в соответствии с инструкцией по его применению), сущность и тяжесть которой не соответствуют информации о лекарственном препарате, содержащейся в инструкции по его применению;
53) рецепт на лекарственный препарат - письменное назначение лекарственного препарата по установленной форме, выданное медицинским или ветеринарным работником, имеющим на это право, в целях отпуска лекарственного препарата или его изготовления и отпуска;
54) требование медицинской организации, ветеринарной организации - документ установленной формы, который выписан медицинским или ветеринарным работником, имеющим на это право, и содержит в письменной форме указание аптечной организации об отпуске лекарственного препарата или о его изготовлении и об отпуске для обеспечения лечебного процесса в медицинской организации, ветеринарной организации.
Глава 4. ГОСУДАРСТВЕННЫЙ КОНТРОЛЬ ПРИ ОБРАЩЕНИИ
ЛЕКАРСТВЕННЫХ СРЕДСТВ
Статья 8. Лицензирование производства лекарственных средств и фармацевтической деятельности.
1. Лицензирование производства лекарственных средств и фармацевтической деятельности осуществляется в соответствии с законодательством Российской Федерации.
2. Обязательным условием предоставления лицензии на производство лекарственных средств является приложение к заявлению соискателя лицензии перечня лекарственных форм и (или) видов фармацевтических субстанций, которые производитель лекарственных средств намерен производить.
3. В случае необходимости расширения производства лекарственных средств за счет новых лекарственных форм и видов фармацевтических субстанций производитель лекарственных средств должен получить новую лицензию на производство лекарственных средств.
Статья 9. Государственный контроль (надзор) в сфере обращения лекарственных средств.
1. Государственный контроль (надзор) в сфере обращения лекарственных средств включает в себя:
1) лицензионный контроль в сфере производства лекарственных средств и в сфере фармацевтической деятельности;
2) федеральный государственный надзор в сфере обращения лекарственных средств.
2. Лицензионный контроль в сфере производства лекарственных средств и в сфере фармацевтической деятельности осуществляется уполномоченными федеральным органом исполнительной власти и органами исполнительной власти субъектов Российской Федерации согласно их компетенции в порядке, установленном Федеральным законом от 26 декабря 2008 года N 294-ФЗ "О защите прав юридических лиц и индивидуальных предпринимателей при осуществлении государственного контроля (надзора) и муниципального контроля", с учетом особенностей организации и проведения проверок, установленных Федеральным законом от 4 мая 2011 года N 99-ФЗ "О лицензировании отдельных видов деятельности".
3. Федеральный государственный надзор в сфере обращения лекарственных средств осуществляется уполномоченными федеральными органами исполнительной власти (далее - органы государственного надзора) согласно их компетенции в порядке, установленном Правительством Российской Федерации.
4. Федеральный государственный надзор в сфере обращения лекарственных средств включает в себя:
1) организацию и проведение проверок соблюдения субъектами обращения лекарственных средств установленных настоящим Федеральным законом и принятыми в соответствии с ним иными нормативными правовыми актами Российской Федерации требований к доклиническим исследованиям лекарственных средств, клиническим исследованиям лекарственных препаратов, хранению, перевозке, ввозу в Российскую Федерацию, отпуску, реализации лекарственных средств, применению лекарственных препаратов, уничтожению лекарственных средств, а также соблюдения уполномоченными органами исполнительной власти субъектов Российской Федерации методики установления предельных размеров оптовых надбавок и предельных размеров розничных надбавок к фактическим отпускным ценам, установленным производителями лекарственных препаратов, на лекарственные препараты, включенные в перечень жизненно необходимых и важнейших лекарственных препаратов (далее - обязательные требования);
2) организацию и проведение проверок соответствия лекарственных средств, находящихся в обращении, установленным обязательным требованиям к их качеству;
3) выдачу разрешений на ввоз лекарственных средств на территорию Российской Федерации;
4) организацию и проведение мониторинга безопасности лекарственных препаратов;
5) применение в порядке, установленном законодательством Российской Федерации, мер по пресечению выявленных нарушений обязательных требований и (или) устранению последствий таких нарушений, выдачу предписаний об устранении выявленных нарушений обязательных требований и привлечение к ответственности лиц, совершивших такие нарушения.
5. К отношениям, связанным с осуществлением федерального государственного надзора в сфере обращения лекарственных средств, организацией и проведением проверок субъектов обращения лекарственных средств, применяются положения Федерального закона от 26 декабря 2008 года N 294-ФЗ "О защите прав юридических лиц и индивидуальных предпринимателей при осуществлении государственного контроля (надзора) и муниципального контроля".
6. Должностные лица органа государственного надзора в порядке, установленном законодательством Российской Федерации, имеют право:
1) получать на основании мотивированных письменных запросов от субъектов обращения лекарственных средств, органов исполнительной власти субъектов Российской Федерации и органов местного самоуправления документы и информацию по вопросам обращения лекарственных средств;
2) беспрепятственно по предъявлении служебного удостоверения и копии приказа (распоряжения) органа государственного надзора о назначении проверки посещать используемые юридическими лицами, индивидуальными предпринимателями, являющимися субъектами обращения лекарственных средств, при осуществлении своей деятельности территории, здания, помещения и сооружения в целях проведения мероприятий по контролю;
3) проводить отбор образцов лекарственных средств, предназначенных для реализации и реализуемых субъектами обращения лекарственных средств, для проверки их качества, проведения исследований, испытаний в соответствии с правилами отбора образцов, установленными уполномоченным федеральным органом исполнительной власти;
4) выдавать субъектам обращения лекарственных средств предписания о прекращении нарушений обязательных требований и об устранении выявленных нарушений обязательных требований;
5) направлять в уполномоченные органы материалы, связанные с нарушениями обязательных требований, для решения вопросов о возбуждении уголовных дел по признакам преступлений.
Глава 6. ОСУЩЕСТВЛЕНИЕ ГОСУДАРСТВЕННОЙ РЕГИСТРАЦИИ
ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ
Статья 13. Государственная регистрация лекарственных препаратов
1. Лекарственные препараты вводятся в гражданский оборот на территории Российской Федерации, если они зарегистрированы соответствующим уполномоченным федеральным органом исполнительной власти.
2. Государственной регистрации подлежат:
1) оригинальные лекарственные препараты;
2) воспроизведенные лекарственные препараты;
3) новые комбинации зарегистрированных ранее лекарственных препаратов;
4) лекарственные препараты, зарегистрированные ранее, но произведенные в других лекарственных формах, в новой дозировке.
3. Государственная регистрация лекарственных препаратов для медицинского применения осуществляется по результатам экспертизы лекарственных средств и этической экспертизы возможности проведения клинического исследования лекарственного препарата для медицинского применения (далее - этическая экспертиза). Государственная регистрация лекарственных препаратов для ветеринарного применения осуществляется по результатам экспертизы лекарственных средств для ветеринарного применения.
4. Государственная регистрация лекарственных препаратов осуществляется соответствующим уполномоченным федеральным органом исполнительной власти в срок, не превышающий двухсот десяти рабочих дней со дня принятия заявления о государственной регистрации лекарственного препарата. В указанный срок включается время, необходимое для повторного проведения экспертизы лекарственных средств и (или) этической экспертизы в соответствии со статьей 25 настоящего Федерального закона. Срок государственной регистрации лекарственного препарата исчисляется со дня принятия соответствующим уполномоченным федеральным органом исполнительной власти заявления о государственной регистрации лекарственного препарата с приложением необходимых документов по день выдачи регистрационного удостоверения лекарственного препарата. Время проведения клинического исследования лекарственного препарата, а также время, необходимое для направления уполномоченным федеральным органом исполнительной власти запроса о представлении необходимых материалов и представления заявителем ответа на данный запрос в соответствии со статьями 16, 19, 22 и 23 настоящего Федерального закона, не учитывается при исчислении срока государственной регистрации такого лекарственного препарата.
5. Государственной регистрации не подлежат:
1) лекарственные препараты, изготовленные аптечными организациями, ветеринарными аптечными организациями, индивидуальными предпринимателями, которые имеют лицензию на фармацевтическую деятельность, по рецептам на лекарственные препараты и требованиям медицинских организаций, ветеринарных организаций;
2) лекарственное растительное сырье;
3) лекарственные препараты, приобретенные физическими лицами за пределами территории Российской Федерации и предназначенные для личного использования;
4) лекарственные препараты, предназначенные для экспорта;
5) радиофармацевтические лекарственные препараты, изготовленные непосредственно в медицинских организациях в порядке, установленном уполномоченным федеральным органом исполнительной власти.
6. Не допускается государственная регистрация:
1) различных лекарственных препаратов под одинаковым торговым наименованием;
2) одного лекарственного препарата, выпускаемого производителем под различными торговыми наименованиями и представленного на государственную регистрацию в виде двух и более лекарственных препаратов.
Статья 14. Принципы экспертизы лекарственных средств и этической экспертизы
1. Экспертиза лекарственных средств и этическая экспертиза основываются на принципах законности, соблюдения прав и свобод человека и гражданина, прав юридического лица, независимости эксперта, объективности, всесторонности и полноты исследований, проводимых с использованием современных достижений науки и техники, ответственности федерального государственного бюджетного учреждения по проведению экспертизы лекарственных средств и экспертов за проведение и качество экспертизы.
2. Экспертиза лекарственных средств для медицинского применения проводится поэтапно:
1) на первом этапе - экспертиза документов для получения разрешения на проведение клинического исследования лекарственного препарата, за исключением:
а) лекарственных препаратов, которые разрешены для медицинского применения на территории Российской Федерации более двадцати лет и в отношении которых невозможно проведение исследования биоэквивалентности;
б) лекарственных препаратов для медицинского применения, в отношении которых проведены международные многоцентровые клинические исследования, часть из которых проведена на территории Российской Федерации;
2) на втором этапе - экспертиза предложенных методов контроля качества лекарственного средства и качества представленных образцов лекарственного средства с использованием этих методов (далее - экспертиза качества лекарственного средства) и экспертиза отношения ожидаемой пользы к возможному риску применения лекарственного препарата, осуществляемые после проведения его клинического исследования.
3. Экспертиза лекарственных средств для ветеринарного применения проводится одноэтапно и включает в себя экспертизу качества лекарственного средства и экспертизу отношения ожидаемой пользы к возможному риску применения лекарственного препарата.
Статья 15. Федеральное государственное бюджетное учреждение по проведению экспертизы лекарственных средств
Экспертиза лекарственных средств проводится федеральным государственным бюджетным учреждением соответствующего уполномоченного федерального органа исполнительной власти, созданным для обеспечения исполнения полномочий этого федерального органа по выдаче разрешений на проведение клинических исследований лекарственных препаратов и (или) по государственной регистрации лекарственных препаратов (далее - экспертное учреждение).
Статья 16. Организация проведения экспертизы лекарственных средств в целях их государственной регистрации
1. Экспертиза лекарственных средств проводится комиссией экспертов экспертного учреждения, назначенной его руководителем, на основании задания на проведение экспертизы лекарственного средства, выданного уполномоченным федеральным органом исполнительной власти. Руководитель экспертного учреждения обеспечивает надлежащее проведение экспертизы лекарственных средств в соответствии с заданием, выданным уполномоченным федеральным органом исполнительной власти, и организует подготовку сводного заключения этой комиссии. В состав этой комиссии по решению руководителя экспертного учреждения могут быть включены в качестве экспертов лица, не работающие в данном экспертном учреждении, если их специальные знания необходимы для проведения экспертизы и такие эксперты отсутствуют в данном экспертном учреждении.
2. Экспертом по проведению экспертизы лекарственных средств является аттестованный работник экспертного учреждения, который имеет высшее медицинское, фармацевтическое, биологическое, ветеринарное или химическое образование и проводит экспертизу лекарственных средств в порядке исполнения своих должностных обязанностей (далее - эксперт).
3. При проведении экспертизы лекарственных средств эксперт не может находиться в какой-либо зависимости от лица, назначившего эту экспертизу, разработчика лекарственного средства или других заинтересованных в результатах экспертизы лиц.
4. При проведении экспертизы лекарственных средств не допускается истребовать у заявителя либо иных лиц материалы, необходимые для проведения экспертизы. В случае недостаточности представленных эксперту материалов для дачи заключения эксперт ставит вопрос о представлении ему необходимых материалов перед руководителем экспертного учреждения, который обращается с соответствующим запросом в уполномоченный федеральный орган исполнительной власти, выдавший задание на проведение экспертизы лекарственного средства. Указанный федеральный орган исполнительной власти в течение пяти рабочих дней со дня поступления запроса руководителя экспертного учреждения направляет заявителю запрос о представлении необходимых материалов. Данный запрос может быть передан уполномоченному представителю заявителя лично под расписку, направлен по почте заказным письмом или передан в электронной форме по телекоммуникационным каналам связи. В случае направления данного запроса по почте заказным письмом он считается полученным по истечении шести дней с даты направления заказного письма.
4.1. Заявитель обязан представить ответ на запрос уполномоченного федерального органа исполнительной власти в срок, не превышающий девяноста рабочих дней со дня его получения. Уполномоченный федеральный орган исполнительной власти, выдавший задание на проведение экспертизы лекарственного средства, в течение пяти рабочих дней со дня поступления от заявителя ответа на данный запрос направляет такой ответ в экспертное учреждение. В случае непредставления по истечении девяноста рабочих дней заявителем ответа на данный запрос уполномоченный федеральный орган исполнительной власти, выдавший задание на проведение экспертизы лекарственного средства, в течение пяти рабочих дней направляет в экспертное учреждение уведомление о непредставлении заявителем ответа на запрос указанного органа. Время со дня направления запроса экспертного учреждения в уполномоченный федеральный орган исполнительной власти до дня получения экспертным учреждением ответа на запрос или уведомления о непредставлении ответа на запрос не учитывается при исчислении срока проведения экспертизы лекарственного средства.
5. Эксперт при проведении порученной ему руководителем экспертного учреждения экспертизы лекарственного средства обязан:
1) провести полное исследование представленных ему объектов, материалов, дать обоснованное и объективное заключение по поставленным перед ним вопросам или мотивированное заключение о невозможности проведения им экспертизы лекарственного средства, если поставленные вопросы выходят за пределы специальных знаний эксперта, объекты исследований и материалы непригодны или недостаточны для проведения исследований и дачи заключения либо современный уровень развития науки не позволяет ответить на поставленные вопросы;
2) не разглашать сведения, которые стали ему известны в связи с проведением экспертизы лекарственного средства, а также сведения, составляющие государственную, коммерческую или иную охраняемую законом тайну;
3) обеспечить сохранность представленных объектов исследований и материалов.
6. Эксперт не вправе:
1) проводить экспертизу лекарственного средства по обращению непосредственно к нему организаций или физических лиц;
2) самостоятельно собирать материалы для проведения экспертизы лекарственного средства;
3) проводить экспертизу лекарственного средства в качестве негосударственного эксперта.
7. В случае необходимости эксперт вправе ходатайствовать перед руководителем экспертного учреждения о привлечении к проведению экспертизы лекарственного средства других экспертов.
8. Каждый эксперт, входящий в состав комиссии экспертов, которой поручено проведение экспертизы лекарственного средства, независимо и самостоятельно проводит исследования, оценивает результаты, полученные им лично и другими экспертами, и формулирует выводы относительно поставленных вопросов в пределах своих специальных знаний.
9. Результаты экспертизы лекарственного средства оформляются заключением комиссии экспертов. В заключении комиссии экспертов указываются перечень исследований, объем проведенных каждым экспертом исследований, установленные каждым из них факты и сделанные в результате исследований выводы. Эксперт, мнение которого не совпадает с решением комиссии экспертов, вправе выразить свое мнение в письменной форме, которое приобщается к заключению комиссии экспертов.
10. Эксперты, входящие в состав комиссии, предупреждаются об ответственности в соответствии с законодательством Российской Федерации за дачу заключения, содержащего необоснованные или фальсифицированные выводы, о чем они дают подписку.
11. Определение уровня профессиональной подготовки экспертов и аттестация их на право проведения экспертизы лекарственных средств осуществляются экспертно-квалификационными комиссиями в порядке, установленном соответствующим уполномоченным федеральным органом исполнительной власти. Уровень профессиональной подготовки экспертов подлежит пересмотру указанными комиссиями не реже чем один раз в пять лет.
12. Правила проведения экспертизы лекарственных средств и форма заключения комиссии экспертов устанавливаются соответствующим уполномоченным федеральным органом исполнительной власти.
Статья 17. Этическая экспертиза
1. Этическая экспертиза проводится в целях выдачи заключения об этической обоснованности возможности проведения клинического исследования лекарственного препарата для медицинского применения советом по этике, созданным в порядке, установленном уполномоченным федеральным органом исполнительной власти.
2. Экспертами совета по этике могут быть представители медицинских, научных организаций, образовательных организаций высшего образования, а также представители общественных организаций, религиозных организаций и средств массовой информации. Данные эксперты не должны находиться в какой-либо зависимости от разработчиков лекарственных препаратов и других лиц, заинтересованных в результатах этической экспертизы.
3. Оплата услуг экспертов совета по этике осуществляется на основании договора, заключенного между уполномоченным федеральным органом исполнительной власти, которым создан совет по этике, и экспертом совета по этике, за счет бюджетных ассигнований, предусмотренных уполномоченному федеральному органу исполнительной власти, создавшему совет по этике, в федеральном бюджете на соответствующий год на обеспечение его деятельности, и в размерах, установленных Правительством Российской Федерации.
4. Эксперты совета по этике несут ответственность в соответствии с законодательством Российской Федерации.
5. Состав совета по этике, положение об этом совете, порядок его деятельности, требования к квалификации и опыту работы по экспертной оценке научных, медицинских и этических аспектов клинических исследований лекарственных препаратов для медицинского применения, предъявляемые к экспертам совета по этике, порядок организации и проведения этической экспертизы, форма заключения совета по этике устанавливаются уполномоченным федеральным органом исполнительной власти. Число представителей медицинских организаций не может превышать половину от общего числа экспертов совета по этике.
6. Информация о составе совета по этике, планах его работы и текущей деятельности размещается на официальном сайте уполномоченного федерального органа исполнительной власти в сети "Интернет" в установленном им порядке.
Статья 18. Подача и рассмотрение заявлений о государственной регистрации лекарственных препаратов и представление необходимых документов.
1. Для государственной регистрации лекарственного препарата разработчик лекарственного препарата или уполномоченное им другое юридическое лицо (далее - заявитель) представляет в соответствующий уполномоченный федеральный орган исполнительной власти, осуществляющий государственную регистрацию лекарственных препаратов, заявление о государственной регистрации лекарственного препарата, а также в порядке, установленном соответствующим уполномоченным федеральным органом исполнительной власти, необходимые документы, из которых формируется регистрационное досье на лекарственный препарат (далее - регистрационное досье).
2. В заявлении о государственной регистрации лекарственного препарата указываются:
1) наименование и адрес заявителя и (или) производителя лекарственного препарата и адрес места осуществления производства лекарственного препарата;
2) наименование лекарственного препарата (международное непатентованное или химическое и торговое наименования);
3) перечень веществ, входящих в состав лекарственного препарата, с указанием количества каждого из них;
4) лекарственная форма, дозировка, способы введения и применения, срок годности лекарственного препарата;
5) описание фармакологических и фармакодинамических или иммунобиологических свойств лекарственного препарата;
6) заявленная производителем лекарственного препарата предельная отпускная цена на лекарственный препарат, включенный в перечень жизненно необходимых и важнейших лекарственных препаратов, в случае его государственной регистрации;
7) отсутствие необходимости проведения клинического исследования, исследования биоэквивалентности лекарственного препарата, разрешенного для медицинского применения на территории Российской Федерации более двадцати лет, с указанием нормативных правовых актов, подтверждающих данный срок применения.
3. Регистрационное досье формируется из следующих документов:
1) проекты макетов первичной упаковки и вторичной (потребительской) упаковки лекарственного препарата;
2) документ, переведенный на русский язык, подтверждающий соответствие производителя регистрируемого лекарственного препарата требованиям правил организации производства и контроля качества лекарственных средств, выданный компетентным органом страны производителя регистрируемого лекарственного препарата и заверенный в установленном порядке;
3) проект нормативной документации или нормативного документа на лекарственный препарат либо указание соответствующей фармакопейной статьи;
4) схема технологического процесса производства лекарственного препарата, ее описание и (или) схема технологического процесса производства фармацевтической субстанции, ее описание;
5) документ, переведенный на русский язык, подтверждающий соответствие производителя фармацевтической субстанции требованиям правил организации производства и контроля качества лекарственных средств, выданный компетентным органом страны производителя фармацевтической субстанции, заверенный в установленном порядке и содержащий:
а) наименование фармацевтической субстанции (международное непатентованное или химическое и торговое наименования);
б) наименование и адрес производителя фармацевтической субстанции;
в) срок годности фармацевтической субстанции;
6) документ, содержащий сведения о показателях качества фармацевтической субстанции, используемой при производстве лекарственных препаратов;
7) нормативная документация или нормативный документ на фармацевтическую субстанцию либо указание соответствующей фармакопейной статьи;
8) информация об условиях хранения, перевозки лекарственного препарата и иная информация;
9) отчет о результатах доклинического исследования лекарственного средства для медицинского применения, содержащий описание, результаты и статистический анализ результатов данного доклинического исследования;
10) отчет о результатах доклинического исследования лекарственного средства и клинического исследования лекарственного препарата для ветеринарного применения;
11) проект протокола клинического исследования лекарственного препарата для медицинского применения;
12) брошюра исследователя;
13) информационный листок пациента;
14) информация о выплатах и компенсациях пациентам (здоровым добровольцам, больным) (далее - пациенты), привлеченным к проведению клинических исследований лекарственного препарата для медицинского применения, исследований биоэквивалентности и (или) терапевтической эквивалентности;
15) отчет о результатах международных многоцентровых клинических исследований лекарственного препарата для медицинского применения, часть из которых проведена на территории Российской Федерации;
16) проект инструкции по применению лекарственного препарата, содержащий следующие сведения:
а) наименование лекарственного средства (международное непатентованное или химическое и торговое наименования);
б) лекарственная форма с указанием наименований и количественного содержания (активности) фармацевтических субстанций и вспомогательных веществ;
в) фармакотерапевтическая группа лекарственного препарата;
г) показания для применения;
д) противопоказания для применения;
е) режим дозирования, способ введения, при необходимости время приема лекарственного препарата, продолжительность лечения (в том числе у детей до и после одного года);
ж) меры предосторожности при применении;
з) симптомы передозировки, меры по оказанию помощи при передозировке;
и) указание, при необходимости, особенностей действия лекарственного препарата при первом приеме или при его отмене;
к) описание, при необходимости, действий врача (фельдшера), специалиста в области ветеринарии, пациента, владельца животного при пропуске приема одной или нескольких доз лекарственного препарата;
л) возможные побочные действия при применении лекарственного препарата;
м) взаимодействие с другими лекарственными препаратами и (или) пищевыми продуктами, кормами;
н) указание возможности и особенностей медицинского применения лекарственного препарата беременными женщинами, женщинами в период грудного вскармливания, детьми, взрослыми, имеющими хронические заболевания;
о) сведения о возможном влиянии лекарственного препарата для медицинского применения на способность управлять транспортными средствами, механизмами;
п) срок годности и указание на запрет применения лекарственного препарата по истечении срока годности;
р) условия хранения;
с) указание на необходимость хранения лекарственного препарата в местах, недоступных для детей;
т) указание, при необходимости, специальных мер предосторожности при уничтожении неиспользованных лекарственных препаратов;
у) сроки возможного использования продукции животного происхождения после введения животному лекарственного препарата для ветеринарного применения;
ф) наименование, адрес производителя лекарственного препарата и адрес места производства лекарственного препарата;
х) условия отпуска;
(пп. "х" введен Федеральным законом от 29.11.2010 N 313-ФЗ)
17) переведенная на русский язык и заверенная в установленном порядке копия документа, подтверждающего регистрацию лекарственного препарата в случае его регистрации вне пределов Российской Федерации;
18) документы, представляемые в соответствии со статьями 19 - 23 настоящего Федерального закона.
4. По желанию заявителя могут быть представлены отчеты о проведенных в стране заявителя и других странах результатах клинических исследований, исследований биоэквивалентности и (или) терапевтической эквивалентности лекарственного препарата (включая эпидемиологические или эпизоотологические исследования иммунобиологических лекарственных препаратов, предназначенных для иммунологической профилактики и лечения инфекционных заболеваний, в том числе у детей), содержащие описания проведенных исследований лекарственного препарата, их результаты и статистический анализ полученных результатов.
5. К заявлению о государственной регистрации лекарственного препарата прилагаются:
1) документ, подтверждающий уплату государственной пошлины за проведение экспертизы документов для получения разрешений на проведение клинических исследований лекарственного препарата для медицинского применения и этической экспертизы при обращении за государственной регистрацией лекарственного препарата;
2) документ, подтверждающий уплату государственной пошлины за проведение экспертизы качества лекарственного средства и экспертизы отношения ожидаемой пользы к возможному риску применения лекарственного препарата, разрешенного для медицинского применения на территории Российской Федерации более двадцати лет, при государственной регистрации лекарственного препарата;
3) документ, подтверждающий уплату государственной пошлины за проведение экспертизы качества лекарственного средства и экспертизы отношения ожидаемой пользы к возможному риску применения лекарственного препарата для медицинского применения, в отношении которого проведены международные многоцентровые клинические исследования, часть из которых проведена на территории Российской Федерации, при государственной регистрации лекарственного препарата;
4) документ, подтверждающий уплату государственной пошлины за проведение экспертизы качества лекарственного средства и экспертизы отношения ожидаемой пользы к возможному риску применения лекарственного препарата для ветеринарного применения при его государственной регистрации.
6. Не допускаются получение, разглашение, использование в коммерческих целях и в целях государственной регистрации лекарственных препаратов информации о результатах доклинических исследований лекарственных средств и клинических исследований лекарственных препаратов, представленной заявителем для государственной регистрации лекарственных препаратов, без его согласия в течение шести лет с даты государственной регистрации лекарственного препарата.
Несоблюдение запрета, установленного настоящей частью, влечет за собой ответственность в соответствии с законодательством Российской Федерации.
На территории Российской Федерации запрещается обращение лекарственных средств, зарегистрированных с нарушением настоящей части.
Статья 19. Принятие решения о выдаче экспертному учреждению и совету по этике задания на проведение экспертизы лекарственных средств
1. В течение пяти рабочих дней со дня принятия заявления о государственной регистрации лекарственного препарата уполномоченный федеральный орган исполнительной власти проводит проверку полноты и достоверности сведений, содержащихся в представленных заявителем материалах, и принимает решение о выдаче задания на проведение:
1) экспертизы лекарственных средств в части экспертизы документов для получения разрешения на проведение клинического исследования лекарственного препарата для медицинского применения в соответствии с целями, указанными в статье 38 настоящего Федерального закона, и этической экспертизы в отношении лекарственных препаратов, для которых не проводились клинические исследования на территории Российской Федерации, на основании документов, указанных в пунктах 1 - 9, 11 - 14, 17 части 3 и пункте 1 части 5 статьи 18 настоящего Федерального закона;
2) экспертизы лекарственных средств в части экспертизы качества лекарственного средства и экспертизы отношения ожидаемой пользы к возможному риску применения лекарственного препарата для медицинского применения в отношении лекарственных препаратов, разрешенных для медицинского применения на территории Российской Федерации более двадцати лет, на основании документов, указанных в пунктах 1 - 9, 16 части 3 и пункте 2 части 5 статьи 18 настоящего Федерального закона, а также лекарственных препаратов, в отношении которых проведены международные многоцентровые клинические исследования, часть из которых проведена на территории Российской Федерации, на основании документов, указанных в пунктах 1 - 9, 15 - 17 части 3 и пункте 3 части 5 статьи 18 настоящего Федерального закона;
3) экспертизы лекарственного средства в отношении лекарственных препаратов для ветеринарного применения на основании документов, указанных в пунктах 1 - 8, 10, подпунктах "а" - "д", "ж" - "м", "п" - "ф" пункта 16, пункте 17 части 3 и пункте 4 части 5 статьи 18 настоящего Федерального закона.
2. Уполномоченный федеральный орган исполнительной власти уведомляет в письменной форме заявителя о принятом решении или в случае отказа с указанием причин такого отказа.
2.1. В случае выявления недостоверности сведений, содержащихся в представленных заявителем материалах, уполномоченный федеральный орган исполнительной власти направляет заявителю запрос об уточнении указанных сведений. Данный запрос может быть передан уполномоченному представителю заявителя лично под расписку, направлен по почте заказным письмом или передан в электронной форме по телекоммуникационным каналам связи. В случае направления данного запроса по почте заказным письмом он считается полученным по истечении шести дней с даты направления заказного письма.
2.2. Заявитель обязан представить ответ на запрос уполномоченного федерального органа исполнительной власти в срок, не превышающий девяноста рабочих дней со дня получения данного запроса. Срок, указанный в части 1 настоящей статьи, приостанавливается со дня направления заявителю запроса уполномоченного федерального органа исполнительной власти до дня получения им ответа на данный запрос.
3. Основанием для отказа в организации экспертиз, указанных в части 1 настоящей статьи, является представление необходимых для проведения этих экспертиз документов, перечисленных в частях 3 и 5 статьи 18 настоящего Федерального закона, в неполном объеме, непредставление заявителем в установленный срок ответа на указанный в части 2.1 настоящей статьи запрос уполномоченного федерального органа исполнительной власти, а также представление документов, не содержащих исчерпывающего перечня необходимых сведений.
Статья 20. Экспертиза документов для получения разрешения на проведение клинического исследования лекарственного препарата для медицинского применения и этической экспертизы
1. Экспертиза документов для получения разрешения на проведение клинического исследования лекарственного препарата для медицинского применения в соответствии с целями, указанными в статье 38 настоящего Федерального закона, этическая экспертиза, составление комиссией экспертов и советом по этике заключений о возможности или невозможности проведения такого клинического исследования и направление этих заключений в уполномоченный федеральный орган исполнительной власти осуществляются в срок, не превышающий тридцати рабочих дней со дня получения экспертным учреждением задания уполномоченного федерального органа исполнительной власти с приложением необходимых документов, указанных в пунктах 9, 11, 12 части 3 статьи 18 настоящего Федерального закона, документов, указанных в части 4 статьи 18 настоящего Федерального закона и представленных по желанию заявителя, и советом по этике задания уполномоченного федерального органа исполнительной власти с приложением необходимых документов, указанных в пунктах 11 - 14 части 3 статьи 18 настоящего Федерального закона.
2. Документы, содержащиеся в регистрационном досье и поступившие в экспертное учреждение, совет по этике для осуществления их экспертизы в целях получения разрешения на проведение клинического исследования лекарственного препарата для медицинского применения, подлежат возврату в уполномоченный федеральный орган исполнительной власти одновременно с заключениями соответствующих экспертиз.
Статья 21. Получение разрешения на проведение клинического исследования лекарственного препарата для медицинского применения.
1. В срок, не превышающий пяти рабочих дней со дня получения заключений, указанных в статье 20 настоящего Федерального закона, уполномоченный федеральный орган исполнительной власти осуществляет оценку поступивших заключений для определения их соответствия заданиям на проведение соответствующих экспертиз и уведомляет заявителя в письменной форме о результатах проведенных экспертиз и о возможности или невозможности выдачи заявителю разрешения на проведение клинического исследования лекарственного препарата для медицинского применения.
2. При принятии решения о возможности выдачи разрешения на проведение клинического исследования лекарственного препарата для медицинского применения уполномоченный федеральный орган исполнительной власти приостанавливает проведение государственной регистрации лекарственного препарата до дня подачи заявителем в уполномоченный федеральный орган исполнительной власти заявления о получении разрешения на проведение клинического исследования лекарственного препарата для медицинского применения.
3. В случае принятия решения о невозможности выдачи разрешения на проведение клинического исследования лекарственного препарата для медицинского применения уполномоченный федеральный орган исполнительной власти прекращает процедуру государственной регистрации лекарственного препарата.
Статья 22. Решение о проведении клинического исследования лекарственного препарата для медицинского применения
1. Для получения разрешения на проведение клинического исследования лекарственного препарата для медицинского применения заявитель представляет в уполномоченный федеральный орган исполнительной власти:
1) заявление о получении разрешения на проведение данного клинического исследования;
2) сведения об опыте работы исследователей по соответствующим специальностям и их опыте работы по проведению клинических исследований;
3) копию договора обязательного страхования жизни, здоровья пациентов, участвующих в клиническом исследовании лекарственного препарата для медицинского применения (далее - договор обязательного страхования), заключенного в соответствии с типовыми правилами обязательного страхования жизни, здоровья пациента, участвующего в клиническом исследовании лекарственного препарата для медицинского применения, утвержденными Правительством Российской Федерации (далее - типовые правила обязательного страхования), с указанием предельной численности пациентов, участвующих в клиническом исследовании лекарственного препарата для медицинского применения;
4) сведения о медицинских организациях, в которых предполагается проведение клинических исследований лекарственного препарата для медицинского применения (полное и сокращенное наименования, организационно-правовая форма, место нахождения и место осуществления деятельности, телефон, телефакс, адрес электронной почты медицинской организации);
5) предполагаемые сроки проведения клинического исследования лекарственного препарата для медицинского применения.
2. В срок, не превышающий пяти рабочих дней со дня принятия указанного в части 1 настоящей статьи заявления с приложением необходимых документов, уполномоченный федеральный орган исполнительной власти:
1) проводит проверку полноты и достоверности сведений, содержащихся в представленных заявителем материалах;
2) принимает решение о выдаче разрешения на проведение клинического исследования лекарственного препарата для медицинского применения или об отказе в выдаче указанного разрешения;
3) уведомляет в письменной форме заявителя о принятом решении или в случае отказа с указанием причин такого отказа;
4) выдает разрешение на проведение клинического исследования лекарственного препарата для медицинского применения в порядке, установленном уполномоченным федеральным органом исполнительной власти.
2.1. В случае выявления недостоверности сведений, содержащихся в представленных заявителем материалах, уполномоченный федеральный орган исполнительной власти направляет заявителю запрос об уточнении указанных сведений. Данный запрос может быть передан уполномоченному представителю заявителя лично под расписку, направлен по почте заказным письмом или передан в электронной форме по телекоммуникационным каналам связи. В случае направления данного запроса по почте заказным письмом он считается полученным по истечении шести дней с даты направления заказного письма.
2.2. Заявитель обязан представить ответ на запрос уполномоченного федерального органа исполнительной власти в срок, не превышающий девяноста рабочих дней со дня получения данного запроса. Срок, указанный в части 2 настоящей статьи, приостанавливается со дня направления заявителю запроса уполномоченного федерального органа исполнительной власти до дня получения им ответа на данный запрос.
3. Основаниями для отказа в выдаче разрешения на проведение клинического исследования лекарственного препарата для медицинского применения являются непредставление документов, указанных в части 1 настоящей статьи, несоответствие содержания представленных документов требованиям настоящего Федерального закона, непредставление заявителем в установленный срок ответа на указанный в части 2.1 настоящей статьи запрос уполномоченного федерального органа исполнительной власти либо наличие заключения комиссии экспертов или заключения совета по этике о невозможности проведения клинического исследования лекарственного препарата для медицинского применения по результатам проведенных экспертиз, предусмотренных статьей 20 настоящего Федерального закона.
Статья 23. Экспертиза качества лекарственного средства и экспертиза отношения ожидаемой пользы к возможному риску применения лекарственного препарата для медицинского применения
1. Экспертиза качества лекарственного средства и экспертиза отношения ожидаемой пользы к возможному риску применения лекарственного препарата для медицинского применения, составление комиссиями экспертов заключений по результатам проведенных экспертиз и направление таких заключений в уполномоченный федеральный орган исполнительной власти осуществляются в срок, не превышающий ста десяти рабочих дней со дня получения экспертным учреждением соответствующего задания уполномоченного федерального органа исполнительной власти с документами, указанными в пунктах 1 - 8, 15 - 17 части 3 статьи 18 настоящего Федерального закона, и отчета о проведенном клиническом исследовании лекарственного препарата для медицинского применения.
2. Для проведения указанных в части 1 настоящей статьи экспертиз заявитель представляет в уполномоченный федеральный орган исполнительной власти:
1) заявление о возобновлении государственной регистрации лекарственного препарата и проведении указанных в части 1 настоящей статьи экспертиз;
2) отчет о проведенном клиническом исследовании лекарственного препарата для медицинского применения;
3) документ, подтверждающий уплату государственной пошлины за проведение экспертизы качества лекарственного средства и экспертизы отношения ожидаемой пользы к возможному риску применения лекарственного препарата для медицинского применения при его государственной регистрации.
3. В срок, не превышающий пяти рабочих дней со дня принятия заявления с документами, указанными в части 1 и пунктах 2 и 3 части 2 настоящей статьи, уполномоченный федеральный орган исполнительной власти:
1) проводит проверку полноты и достоверности данных, содержащихся в представленном заявителем отчете о проведении клинического исследования лекарственного препарата для медицинского применения и в документах, содержащихся в регистрационном досье;
2) принимает решение о возобновлении государственной регистрации лекарственного препарата и проведении указанных в части 1 настоящей статьи экспертиз или об отказе в возобновлении государственной регистрации лекарственного препарата для медицинского применения и проведении таких экспертиз;
3) уведомляет в письменной форме заявителя о принятом решении или в случае отказа с указанием причин такого отказа.
3.1. В случае выявления недостоверности сведений, содержащихся в представленных заявителем материалах, уполномоченный федеральный орган исполнительной власти направляет заявителю запрос об уточнении указанных сведений. Данный запрос может быть передан уполномоченному представителю заявителя лично под расписку, направлен по почте заказным письмом или передан в электронной форме по телекоммуникационным каналам связи. В случае направления данного запроса по почте заказным письмом он считается полученным по истечении шести дней с даты направления заказного письма.
3.2. Заявитель обязан представить ответ на запрос уполномоченного федерального органа исполнительной власти в срок, не превышающий девяноста рабочих дней со дня получения данного запроса. Срок, указанный в части 3 настоящей статьи, приостанавливается со дня направления заявителю запроса уполномоченного федерального органа исполнительной власти до дня получения им ответа на данный запрос.
4. Основанием для отказа в возобновлении государственной регистрации лекарственного препарата и проведении указанных в части 1 настоящей статьи экспертиз является представление документов, указанных в части 1 и пунктах 2 и 3 части 2 настоящей статьи, в неполном объеме, непредставление заявителем в установленный срок ответа на указанный в части 3.1 настоящей статьи запрос уполномоченного федерального органа исполнительной власти или представление документов, не содержащих исчерпывающего перечня необходимых сведений.
5. В течение пятнадцати рабочих дней со дня получения решения уполномоченного федерального органа исполнительной власти о возобновлении государственной регистрации лекарственного препарата и проведении указанных в части 1 настоящей статьи экспертиз заявитель представляет в экспертное учреждение для проведения экспертизы качества лекарственного средства образцы лекарственного препарата для медицинского применения, произведенного в соответствии с требованиями опытно-промышленного и (или) промышленного регламентов, утвержденных руководителем производителя лекарственных средств, а также в соответствующих случаях образец фармацевтической субстанции, тест-штамма микроорганизмов, культуры клеток, образцы веществ, применяемых для контроля качества лекарственного средства путем сравнения с ними исследуемого лекарственного средства, в количествах, необходимых для воспроизведения методов контроля качества.
6. При получении образцов лекарственного препарата и фармацевтической субстанции экспертное учреждение выдает заявителю документ, подтверждающий получение указанных образцов, и в срок, не превышающий трех рабочих дней, уведомляет в письменной форме об этом уполномоченный федеральный орган исполнительной власти.
7. Срок для представления заявителем образцов лекарственного препарата и фармацевтической субстанции и срок уведомления экспертным учреждением об этом в письменной форме уполномоченного федерального органа исполнительной власти, указанные в частях 5 и 6 настоящей статьи, не включаются в срок проведения указанных в части 1 настоящей статьи экспертиз.
8. Документы, поступившие в экспертное учреждение для проведения указанных в части 1 настоящей статьи экспертиз, подлежат возврату в уполномоченный федеральный орган исполнительной власти одновременно с заключениями по результатам указанных экспертиз.
Статья 24. Экспертиза качества лекарственного средства и экспертиза отношения ожидаемой пользы к возможному риску применения лекарственного препарата для ветеринарного применения
1. Экспертиза качества лекарственного средства и экспертиза отношения ожидаемой пользы к возможному риску применения лекарственного препарата для ветеринарного применения, составление комиссиями экспертов заключений по результатам указанных экспертиз и направление таких заключений в уполномоченный федеральный орган исполнительной власти осуществляются в срок, не превышающий ста десяти рабочих дней со дня получения экспертным учреждением задания уполномоченного федерального органа исполнительной власти с документами, указанными в пунктах 1 - 8, 10, подпунктах "а" - "д", "ж" - "м", "п" - "ф" пункта 16 и пункте 17 части 3 статьи 18 настоящего Федерального закона.
2. В течение пятнадцати рабочих дней со дня получения решения уполномоченного федерального органа исполнительной власти о проведении указанных в части 1 настоящей статьи экспертиз заявитель представляет в экспертное учреждение для проведения экспертизы качества лекарственного средства образцы лекарственного препарата для ветеринарного применения, произведенного в соответствии с требованиями технологических регламентов, утвержденных руководителем производителя лекарственных средств, а также образец фармацевтической субстанции в количествах, необходимых для воспроизведения методов контроля качества.
3. При получении образцов лекарственного препарата и фармацевтической субстанции экспертное учреждение выдает заявителю документ, подтверждающий получение указанных образцов, и в срок, не превышающий трех рабочих дней, уведомляет в письменной форме об этом уполномоченный федеральный орган исполнительной власти.
4. Срок для представления заявителем образцов лекарственного препарата и фармацевтической субстанции и срок уведомления экспертным учреждением об этом в письменной форме уполномоченного федерального органа исполнительной власти, указанные в частях 2 и 3 настоящей статьи, не включаются в срок проведения указанных в части 1 настоящей статьи экспертиз.
5. Документы, поступившие в экспертное учреждение для проведения указанных в части 1 настоящей статьи экспертиз, подлежат возврату в уполномоченный федеральный орган исполнительной власти одновременно с заключениями по результатам указанных экспертиз.
Статья 25. Повторное проведение экспертизы лекарственных средств и этической экспертизы.
1. В случаях недостаточной обоснованности или неполноты заключения комиссии экспертов или совета по этике, наличия в нем противоречивых данных, фальсификации выводов экспертизы лекарственного средства и (или) этической экспертизы, сокрытия от уполномоченного федерального органа исполнительной власти оснований для отвода эксперта вследствие его заинтересованности в результатах соответствующей экспертизы, наличия данных о прямом либо косвенном вмешательстве в процесс соответствующей экспертизы лиц, не участвующих в ее проведении, но оказавших влияние на процесс и результаты ее проведения, уполномоченным федеральным органом исполнительной власти назначается повторная экспертиза лекарственного средства и (или) этическая экспертиза.
2. Повторная экспертиза лекарственного средства проводится в срок, установленный уполномоченным федеральным органом исполнительной власти и не превышающий сорока рабочих дней со дня получения экспертным учреждением задания на проведение повторной экспертизы лекарственного средства, повторная этическая экспертиза - в срок, не превышающий пятнадцати рабочих дней со дня получения советом по этике задания на проведение повторной этической экспертизы.
3. Финансовое обеспечение выполнения задания на проведение повторной экспертизы лекарственного средства не осуществляется, и средства, перечисленные ранее на проведение такой экспертизы, подлежат возврату в федеральный бюджет.
Статья 26. Ускоренная процедура экспертизы лекарственных средств
1. Ускоренная процедура экспертизы лекарственных средств в целях государственной регистрации лекарственных препаратов применяется в отношении воспроизведенных лекарственных препаратов. При проведении такой процедуры представляются информация, полученная при проведении клинических исследований лекарственных препаратов и опубликованная в специализированных печатных изданиях, а также документы, содержащие результаты исследования биоэквивалентности и (или) терапевтической эквивалентности лекарственного препарата для медицинского применения или результаты исследования биоэквивалентности лекарственного препарата для ветеринарного применения.
2. Ускоренная процедура экспертизы лекарственных средств не применяется в отношении иммунобиологических лекарственных препаратов, препаратов инсулина и лекарственных препаратов, впервые регистрируемых в Российской Федерации.
3. Ускоренная процедура экспертизы лекарственных средств проводится по решению соответствующего уполномоченного федерального органа исполнительной власти в срок, не превышающий шестидесяти рабочих дней. При этом экспертиза документов, содержащихся в регистрационном досье, для получения разрешения на проведение клинического исследования лекарственного препарата для медицинского применения и этическая экспертиза проводятся в срок, не превышающий пятнадцати рабочих дней, экспертиза качества лекарственного средства и экспертиза отношения ожидаемой пользы к возможному риску применения лекарственного препарата - в срок, не превышающий сорока пяти рабочих дней.
4. Ускоренная процедура экспертизы лекарственных средств проводится в порядке, установленном статьями 17 - 20, 23 и 24 настоящего Федерального закона, и не означает снижения требований к безопасности, качеству и эффективности лекарственных препаратов.
Статья 27. Решение о государственной регистрации лекарственного препарата
1. В срок, не превышающий пяти рабочих дней со дня получения заключений комиссии экспертов по результатам экспертизы качества лекарственного средства и экспертизы отношения ожидаемой пользы к возможному риску применения лекарственного препарата, соответствующий уполномоченный федеральный орган исполнительной власти:
1) осуществляет оценку таких заключений для определения их соответствия заданию на проведение указанных экспертиз;
2) принимает решение о государственной регистрации лекарственного препарата или об отказе в государственной регистрации лекарственного препарата;
3) вносит при принятии решения о государственной регистрации лекарственного препарата данные о зарегистрированном лекарственном препарате, в том числе о фармацевтической субстанции, входящей в состав лекарственного препарата, в государственный реестр лекарственных средств и выдает заявителю регистрационное удостоверение лекарственного препарата, форма которого утверждается уполномоченным федеральным органом исполнительной власти, согласованные нормативную документацию, нормативный документ, инструкцию по применению лекарственного препарата и макеты первичной упаковки и вторичной (потребительской) упаковки с указанием на них номера регистрационного удостоверения лекарственного препарата и даты его государственной регистрации или в случае принятия решения об отказе в государственной регистрации лекарственного препарата уведомляет в письменной форме заявителя об этом с указанием причин такого отказа.
2. Основанием для отказа в государственной регистрации лекарственного препарата является решение соответствующего уполномоченного федерального органа исполнительной власти о том, что качество и (или) эффективность регистрируемого лекарственного препарата не подтверждены полученными данными или что риск причинения вреда здоровью человека или животного вследствие приема лекарственного препарата превышает эффективность его применения.
3. При государственной регистрации лекарственного препарата, включенного в перечень жизненно необходимых и важнейших лекарственных препаратов, необходимые данные заносятся в государственный реестр предельных отпускных цен производителей на лекарственные препараты, включенные в указанный перечень.
Статья 28. Регистрационное удостоверение лекарственного препарата
1. Регистрационное удостоверение лекарственного препарата с указанием лекарственных форм и дозировок выдается бессрочно, за исключением регистрационного удостоверения лекарственного препарата, выдаваемого со сроком действия пять лет, на впервые регистрируемые в Российской Федерации лекарственные препараты.
2. По истечении указанного в части 1 настоящей статьи срока выдается бессрочное регистрационное удостоверение лекарственного препарата при условии подтверждения его государственной регистрации.
Статья 29. Подтверждение государственной регистрации лекарственного препарата
1. Подтверждение государственной регистрации лекарственного препарата осуществляется при выдаче бессрочного регистрационного удостоверения лекарственного препарата в случае, указанном в части 2 статьи 28 настоящего Федерального закона, в срок, не превышающий девяноста рабочих дней со дня получения соответствующим уполномоченным федеральным органом исполнительной власти заявления о подтверждении государственной регистрации лекарственного препарата, оформленного в соответствии с частью 2 статьи 18 настоящего Федерального закона.
2. Подтверждение государственной регистрации лекарственного препарата осуществляется по результатам экспертизы отношения ожидаемой пользы к возможному риску применения лекарственного препарата, а также экспертизы качества лекарственного средства, проводимой в случае внесения изменений в нормативную документацию или нормативный документ.
3. К заявлению о подтверждении государственной регистрации лекарственного препарата прилагаются документ, подтверждающий уплату государственной пошлины за подтверждение государственной регистрации лекарственного препарата для медицинского применения или лекарственного препарата для ветеринарного применения, документ, содержащий результаты мониторинга безопасности лекарственного препарата, проводимого заявителем, по форме, установленной соответствующим уполномоченным федеральным органом исполнительной власти. Нормативная документация или нормативный документ, проект инструкции по применению лекарственного препарата, проекты макетов первичной упаковки и вторичной (потребительской) упаковки лекарственного препарата прилагаются к заявлению о подтверждении государственной регистрации лекарственного препарата вновь только в случае, если в них вносятся изменения.
4. В течение десяти рабочих дней со дня принятия заявления о подтверждении государственной регистрации лекарственного препарата и необходимых документов соответствующий уполномоченный федеральный орган исполнительной власти:
1) проводит проверку полноты и достоверности сведений, содержащихся в представленных заявителем материалах;
2) принимает решение о проведении или об отказе в проведении экспертизы отношения ожидаемой пользы к возможному риску применения лекарственного препарата, а также экспертизы качества лекарственного средства, проводимой в случае внесения изменений в нормативную документацию или нормативный документ;
3) уведомляет в письменной форме заявителя о принятом решении или в случае принятия решения об отказе в проведении экспертизы с указанием причин такого отказа.
4.1. В случае выявления недостоверности сведений, содержащихся в представленных заявителем материалах, уполномоченный федеральный орган исполнительной власти направляет заявителю запрос об уточнении указанных сведений. Данный запрос может быть передан уполномоченному представителю заявителя лично под расписку, направлен по почте заказным письмом или передан в электронной форме по телекоммуникационным каналам связи. В случае направления данного запроса по почте заказным письмом он считается полученным по истечении шести дней с даты направления заказного письма.
4.2. Заявитель обязан представить ответ на запрос уполномоченного федерального органа исполнительной власти в срок, не превышающий девяноста рабочих дней со дня получения данного запроса. Срок, указанный в части 4 настоящей статьи, приостанавливается со дня направления заявителю запроса уполномоченного федерального органа исполнительной власти до дня получения им ответа на данный запрос и не учитывается при исчислении срока подтверждения государственной регистрации лекарственного препарата.
5. Основанием для отказа в проведении экспертизы отношения ожидаемой пользы к возможному риску применения лекарственного препарата и (или) экспертизы качества лекарственного средства является представление документов, указанных в частях 1 и 3 настоящей статьи, в неполном объеме, непредставление заявителем в установленный срок ответа на указанный в части 4.1 настоящей статьи запрос уполномоченного федерального органа исполнительной власти, а также отсутствие в представленных документах исчерпывающих сведений, которые должны быть отражены в них.
6. Экспертиза отношения ожидаемой пользы к возможному риску применения лекарственного препарата и (или) экспертиза качества лекарственного средства в целях подтверждения государственной регистрации лекарственного препарата проводятся на основании документов, указанных в части 3 настоящей статьи, в порядке, установленном частями 5 - 8 статьи 23 и статьей 24 настоящего Федерального закона.
7. В период проведения процедуры подтверждения государственной регистрации лекарственного препарата его гражданский оборот осуществляется на территории Российской Федерации.
Статья 30. Внесение изменений в документы, содержащиеся в регистрационном досье на зарегистрированный лекарственный препарат для медицинского применения
1. В целях внесения изменений в документы, содержащиеся в регистрационном досье на зарегистрированный лекарственный препарат для медицинского применения, заявитель представляет в уполномоченный федеральный орган исполнительной власти заявление о внесении таких изменений по форме, установленной уполномоченным федеральным органом исполнительной власти, и приложенные к нему изменения в указанные документы, а также документы, подтверждающие необходимость внесения таких изменений. Принятие решения о внесении таких изменений или об отказе в их внесении осуществляется в срок, не превышающий девяноста рабочих дней со дня принятия уполномоченным федеральным органом исполнительной власти заявления о внесении таких изменений.
2. В случае внесения изменений в инструкцию по применению лекарственного препарата в отношении сведений, указанных в подпунктах "г" - "п", "х" пункта 16 части 3 статьи 18 настоящего Федерального закона, в состав лекарственного препарата для медицинского применения, изменения места производства лекарственного препарата для медицинского применения, изменения показателей качества лекарственного препарата для медицинского применения и (или) методов контроля качества лекарственного препарата для медицинского применения, изменения срока годности лекарственного препарата для медицинского применения проводится экспертиза лекарственных средств в части экспертизы качества лекарственного средства и (или) экспертизы отношения ожидаемой пользы к возможному риску применения лекарственного препарата для медицинского применения. В случае необходимости внесения иных изменений в данную инструкцию экспертиза лекарственного средства для внесения таких изменений в сведения о зарегистрированном лекарственном препарате не проводится.
3. С заявлением о внесении изменений в документы, содержащиеся в регистрационном досье на зарегистрированный лекарственный препарат для медицинского применения, наряду с документами, указанными в части 1 настоящей статьи, представляются документы, подтверждающие уплату государственной пошлины за внесение изменений в инструкцию по применению лекарственного препарата для медицинского применения, государственной пошлины за внесение изменений в состав лекарственного препарата для медицинского применения.
4. В течение десяти рабочих дней со дня поступления указанного в части 1 настоящей статьи заявления и необходимых документов уполномоченный федеральный орган исполнительной власти:
1) проводит проверку полноты и достоверности сведений, содержащихся в представленных заявителем материалах;
2) принимает решение о проведении указанных в части 2 настоящей статьи соответствующих экспертиз лекарственного средства или об отказе в их проведении;
3) уведомляет в письменной форме заявителя о принятом решении или в случае принятия решения об отказе в проведении соответствующей экспертизы с указанием причин такого отказа.
4.1. В случае выявления недостоверности сведений, содержащихся в представленных заявителем материалах, уполномоченный федеральный орган исполнительной власти направляет заявителю запрос об уточнении указанных сведений. Данный запрос может быть передан уполномоченному представителю заявителя лично под расписку, направлен по почте заказным письмом или передан в электронной форме по телекоммуникационным каналам связи. В случае направления данного запроса по почте заказным письмом он считается полученным по истечении шести дней с даты направления заказного письма.
4.2. Заявитель обязан представить ответ на запрос уполномоченного федерального органа исполнительной власти в срок, не превышающий девяноста рабочих дней со дня получения данного запроса. Срок, указанный в части 4 настоящей статьи, приостанавливается со дня направления заявителю запроса уполномоченного федерального органа исполнительной власти до дня получения им ответа на данный запрос и не учитывается при исчислении срока принятия решения о внесении изменений в документы, содержащиеся в регистрационном досье на зарегистрированный лекарственный препарат для медицинского применения.
5. Основанием для отказа в проведении указанных в части 2 настоящей статьи экспертиз является представление документов, перечисленных в частях 1 и 3 настоящей статьи, в неполном объеме, непредставление заявителем в установленный срок ответа на указанный в части 4.1 настоящей статьи запрос уполномоченного федерального органа исполнительной власти, а также отсутствие в представленных документах достаточных сведений, подтверждающих необходимость внесения изменений.
6. Указанные в части 2 настоящей статьи экспертизы проводятся в порядке, установленном статьей 23 настоящего Федерального закона.
7. В срок, не превышающий пяти рабочих дней со дня получения заключений комиссий экспертов по результатам указанных в части 2 настоящей статьи экспертиз, уполномоченный федеральный орган исполнительной власти:
1) принимает решение о внесении изменений в документы, содержащиеся в регистрационном досье на зарегистрированный лекарственный препарат для медицинского применения, или об отказе во внесении таких изменений;
2) вносит в государственный реестр лекарственных средств на основании решения о внесении изменений в документы, содержащиеся в регистрационном досье на зарегистрированный лекарственный препарат для медицинского применения, необходимые изменения и возвращает их заявителю.
8. Основанием для отказа во внесении изменений в документы, содержащиеся в регистрационном досье на зарегистрированный лекарственный препарат для медицинского применения, является заключение уполномоченного федерального органа исполнительной власти о возможности снижения безопасности, качества, эффективности лекарственного средства в случае внесения таких изменений.
9. Допускается гражданский оборот лекарственных препаратов для медицинского применения, произведенных до принятия уполномоченным федеральным органом исполнительной власти решения о внесении изменений в документы, содержащиеся в регистрационном досье на такие лекарственные препараты, или решения об отказе во внесении указанных изменений.
Статья 31. Внесение изменений в документы, содержащиеся в регистрационном досье на зарегистрированный лекарственный препарат для ветеринарного применения
1. В целях внесения изменений в документы, содержащиеся в регистрационном досье на зарегистрированный лекарственный препарат для ветеринарного применения, заявитель представляет в уполномоченный федеральный орган исполнительной власти заявление о внесении таких изменений по форме, установленной уполномоченным федеральным органом исполнительной власти, и приложенные к нему изменения в указанные документы, а также документы, подтверждающие необходимость внесения таких изменений. Принятие решения о внесении таких изменений или об отказе в их внесении осуществляется в срок, не превышающий девяноста рабочих дней со дня принятия уполномоченным федеральным органом исполнительной власти заявления о внесении таких изменений.
2. В случае внесения изменений в инструкцию по применению лекарственного препарата для ветеринарного применения в отношении сведений об изменениях дозировки, сроков возможного использования продукции животного происхождения после применения лекарственного препарата для ветеринарного применения проводится экспертиза лекарственного средства для ветеринарного применения. В случае необходимости внесения иных изменений в данную инструкцию экспертиза лекарственного средства для ветеринарного применения не проводится.
3. С заявлением о внесении изменений в документы, содержащиеся в регистрационном досье на зарегистрированный лекарственный препарат для ветеринарного применения, наряду с документами, указанными в части 1 настоящей статьи, представляются документы, подтверждающие уплату государственной пошлины за внесение изменений в инструкцию по применению лекарственного препарата для ветеринарного применения.
4. В срок, не превышающий десяти рабочих дней со дня поступления указанного в части 1 настоящей статьи заявления и необходимых документов, уполномоченный федеральный орган исполнительной власти:
1) проводит проверку полноты и достоверности сведений, содержащихся в представленных заявителем документах;
2) принимает решение о проведении экспертизы лекарственного средства для ветеринарного применения или об отказе в ее проведении;
3) уведомляет в письменной форме заявителя о принятом решении или в случае принятия решения об отказе в проведении экспертизы лекарственного средства для ветеринарного применения с указанием причин такого отказа.
5. Основанием для отказа в проведении экспертизы лекарственного средства для ветеринарного применения является представление документов, указанных в частях 1 и 3 настоящей статьи, в неполном объеме или отсутствие в представленных документах достаточных сведений, подтверждающих необходимость внесения изменений.
6. Экспертиза лекарственного средства для ветеринарного применения в целях внесения изменений в документы, содержащиеся в регистрационном досье на зарегистрированный лекарственный препарат для ветеринарного применения, осуществляется в порядке, установленном статьей 24 настоящего Федерального закона.
7. Основанием для отказа во внесении изменений в документы, содержащиеся в регистрационном досье на зарегистрированный лекарственный препарат для ветеринарного применения, является заключение уполномоченного федерального органа исполнительной власти о возможности снижения безопасности, качества, эффективности лекарственного препарата для ветеринарного применения в случае внесения изменений в указанные документы.
8. Допускается гражданский оборот лекарственных препаратов для ветеринарного применения, произведенных до принятия уполномоченным федеральным органом исполнительной власти решения о внесении изменений в документы, содержащиеся в регистрационном досье на зарегистрированный лекарственный препарат для ветеринарного применения, или об отказе во внесении указанных изменений.
Статья 32. Отмена государственной регистрации лекарственного препарата
Решение об отмене государственной регистрации лекарственного препарата и исключении лекарственного препарата из государственного реестра лекарственных средств принимается уполномоченным федеральным органом исполнительной власти в случае:
1) представления соответствующим уполномоченным федеральным органом исполнительной власти заключения о риске или об угрозе здоровью, жизни человека или животного при применении лекарственного препарата, превышающих его эффективность, по результатам осуществляемого им мониторинга безопасности лекарственного препарата;
2) подачи разработчиком лекарственного средства или уполномоченным им другим юридическим лицом заявления об отмене государственной регистрации лекарственного препарата;
3) неподтверждения государственной регистрации лекарственного препарата по истечении срока действия регистрационного удостоверения, выданного на пять лет;
4) непредставления заявителем информации, которая может повлечь за собой необходимость внесения изменений в документы, содержащиеся в регистрационном досье на зарегистрированный лекарственный препарат, в течение тридцати рабочих дней со дня наступления этих изменений;
5) осуществления государственной регистрации лекарственного препарата под торговым наименованием зарегистрированного ранее под этим торговым наименованием лекарственного препарата;
6) осуществления государственной регистрации заявителем одного и того же лекарственного препарата под различными торговыми наименованиями;
7) вынесения судом решения о нарушении прав правообладателя объектов интеллектуальной собственности при обращении лекарственных средств.
Статья 33. Государственный реестр лекарственных средств
1. Государственный реестр лекарственных средств содержит перечень лекарственных препаратов, прошедших государственную регистрацию, перечень фармацевтических субстанций, входящих в состав лекарственных препаратов, и следующую информацию:
1) в отношении лекарственных препаратов:
а) наименование лекарственного препарата (международное непатентованное или химическое и торговое наименования);
б) лекарственная форма с указанием дозировки лекарственного препарата и его количества в потребительской упаковке;
в) наименование разработчика лекарственного препарата;
г) наименование и адрес производителя лекарственного препарата;
д) фармакотерапевтическая группа лекарственного препарата;
е) показания и противопоказания к применению лекарственного препарата;
ж) побочные действия лекарственного препарата;
з) срок годности лекарственного препарата;
и) условия хранения лекарственного препарата;
к) условия отпуска лекарственного препарата;
л) номер фармакопейной статьи или в случае ее отсутствия номер нормативной документации либо нормативного документа;
м) дата государственной регистрации лекарственного препарата и его регистрационный номер;
2) в отношении фармацевтических субстанций:
а) наименование фармацевтической субстанции (международное непатентованное или химическое и торговое наименования);
б) наименование и адрес производителя фармацевтической субстанции;
в) срок годности фармацевтической субстанции;
г) условия хранения фармацевтической субстанции;
д) номер фармакопейной статьи или в случае ее отсутствия номер нормативной документации либо нормативного документа.
2. Фармацевтическая субстанция, неиспользуемая при производстве лекарственных препаратов, может быть включена в государственный реестр лекарственных средств на основании заявления разработчика, производителя лекарственного средства либо уполномоченного ими юридического лица при условии проведения в отношении такой фармацевтической субстанции экспертизы качества фармацевтической субстанции в порядке, установленном статьей 34 настоящего Федерального закона.
3. Порядок ведения государственного реестра лекарственных средств для медицинского применения и порядок ведения государственного реестра лекарственных средств для ветеринарного применения утверждаются соответствующим уполномоченным федеральным органом исполнительной власти.
Статья 34. Экспертиза качества фармацевтической субстанции, неиспользуемой при производстве лекарственных препаратов
1. Для включения фармацевтической субстанции, неиспользуемой при производстве лекарственных препаратов, в государственный реестр лекарственных средств, проводится экспертиза ее качества.
2. Экспертиза качества указанной в части 1 настоящей статьи фармацевтической субстанции, составление комиссией экспертов заключений по результатам такой экспертизы и их направление в уполномоченный федеральный орган исполнительной власти осуществляются в срок, не превышающий шестидесяти рабочих дней со дня получения экспертным учреждением соответствующего задания уполномоченного федерального органа исполнительной власти и документов, указанных в пунктах 4 - 7 части 3 статьи 18 настоящего Федерального закона.
3. Для проведения экспертизы качества указанной в части 1 настоящей статьи фармацевтической субстанции заявитель представляет в уполномоченный федеральный орган исполнительной власти:
1) заявление о включении в государственный реестр лекарственных средств данной фармацевтической субстанции;
2) документ, подтверждающий уплату государственной пошлины за включение фармацевтической субстанции, неиспользуемой при производстве лекарственных препаратов, в государственный реестр лекарственных средств;
3) документы, указанные в пунктах 4 - 7 части 3 статьи 18 настоящего Федерального закона.
4. В течение пяти рабочих дней со дня принятия заявления о включении указанной в части 1 настоящей статьи фармацевтической субстанции в государственный реестр лекарственных средств и документов, перечисленных в части 1 и пункте 2 части 3 настоящей статьи, уполномоченный федеральный орган исполнительной власти:
1) проводит проверку полноты и достоверности данных, содержащихся в представленных заявителем документах;
2) принимает решение о направлении в экспертное учреждение задания на проведение экспертизы качества указанной в части 1 настоящей статьи фармацевтической субстанции или об отказе в таком направлении;
3) уведомляет в письменной форме заявителя о принятом решении или в случае отказа с указанием причин такого отказа.
4.1. В случае выявления недостоверности сведений, содержащихся в представленных заявителем материалах, уполномоченный федеральный орган исполнительной власти направляет заявителю запрос об уточнении указанных сведений. Данный запрос может быть передан уполномоченному представителю заявителя лично под расписку, направлен по почте заказным письмом или передан в электронной форме по телекоммуникационным каналам связи. В случае направления данного запроса по почте заказным письмом он считается полученным по истечении шести дней с даты направления заказного письма.
4.2. Заявитель обязан представить ответ на запрос уполномоченного федерального органа исполнительной власти в срок, не превышающий девяноста рабочих дней со дня получения данного запроса. Срок, указанный в части 4 настоящей статьи, приостанавливается со дня направления заявителю запроса уполномоченного федерального органа исполнительной власти до дня получения им ответа на данный запрос и не учитывается при исчислении срока экспертизы качества фармацевтической субстанции, неиспользуемой при производстве лекарственных препаратов.
5. Основанием для отказа в направлении в экспертное учреждение задания на проведение экспертизы качества указанной в части 1 настоящей статьи фармацевтической субстанции является непредставление документов, перечисленных в части 2 и пункте 2 части 3 настоящей статьи, непредставление заявителем в установленный срок ответа на указанный в части 4.1 настоящей статьи запрос уполномоченного федерального органа исполнительной власти или представление документов, не содержащих исчерпывающего перечня необходимых сведений.
6. В течение пятнадцати рабочих дней со дня получения решения уполномоченного федерального органа исполнительной власти о направлении в экспертное учреждение задания на проведение экспертизы качества указанной в части 1 настоящей статьи фармацевтической субстанции заявитель представляет в экспертное учреждение для проведения данной экспертизы образцы фармацевтической субстанции в количестве, необходимом для воспроизведения методов контроля качества. Экспертное учреждение при получении образцов указанной в части 1 настоящей статьи фармацевтической субстанции выдает заявителю документ, подтверждающий получение этих образцов, и в срок, не превышающий трех рабочих дней, уведомляет в письменной форме об этом уполномоченный федеральный орган исполнительной власти. Эти сроки не включаются в срок проведения данной экспертизы.
7. Документы, поступившие в экспертное учреждение для проведения экспертизы качества указанной в части 1 настоящей статьи фармацевтической субстанции, подлежат возврату в уполномоченный федеральный орган исполнительной власти одновременно с заключениями по результатам проведенной экспертизы.
8. В срок, не превышающий пяти рабочих дней со дня получения заключения комиссии экспертов по результатам экспертизы качества указанной в части 1 настоящей статьи фармацевтической субстанции, уполномоченный федеральный орган исполнительной власти:
1) осуществляет оценку такого заключения для определения его соответствия заданию на проведение данной экспертизы;
2) принимает решение о включении указанной в части 1 настоящей статьи фармацевтической субстанции в государственный реестр лекарственных средств или решение об отказе в таком включении;
3) вносит при принятии решения о включении указанной в части 1 настоящей статьи фармацевтической субстанции в государственный реестр лекарственных средств предусмотренную пунктом 2 части 1 статьи 33 настоящего Федерального закона информацию и уведомляет об этом в письменной форме заявителя.
Статья 35. Повторное представление лекарственного препарата, не прошедшего государственной регистрации лекарственных препаратов, на государственную регистрацию лекарственных препаратов.
Повторное представление в соответствующий уполномоченный федеральный орган исполнительной власти лекарственного препарата, не прошедшего государственной регистрации лекарственных препаратов или получившего отказ в указанной регистрации и впоследствии подвергшегося изменению в части его состава, рассматривается как представление нового лекарственного препарата на его государственную регистрацию независимо от сохранения его первичного наименования.
Статья 36. Обжалование решения об отказе в выдаче разрешения на проведение клинических исследований лекарственного препарата или решения об отказе в государственной регистрации лекарственного препарата
Решение соответствующего уполномоченного федерального органа исполнительной власти об отказе в выдаче разрешения на проведение клинических исследований лекарственного препарата или решение об отказе в государственной регистрации лекарственного препарата может быть обжаловано в порядке, установленном законодательством Российской Федерации.
Статья 37. Информация, связанная с осуществлением государственной регистрации лекарственных препаратов, информация о зарегистрированных лекарственных препаратах и лекарственных препаратах, исключенных из государственного реестра лекарственных средств
1. Соответствующий уполномоченный федеральный орган исполнительной власти размещает на своем официальном сайте в сети "Интернет" информацию, связанную с осуществлением государственной регистрации лекарственных препаратов, в том числе проведением экспертизы лекарственных средств, информацию о зарегистрированных лекарственных препаратах и лекарственных препаратах, исключенных из государственного реестра лекарственных средств, не позднее чем через пять рабочих дней со дня получения соответствующим уполномоченным федеральным органом исполнительной власти заявления о государственной регистрации лекарственного препарата.
2. Сроки и порядок размещения указанной в части 1 настоящей статьи информации устанавливаются уполномоченным федеральным органом исполнительной власти.
Глава 6. ОСУЩЕСТВЛЕНИЕ ГОСУДАРСТВЕННОЙ РЕГИСТРАЦИИ
ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ
Статья 13. Государственная регистрация лекарственных препаратов
1. Лекарственные препараты вводятся в гражданский оборот на территории Российской Федерации, если они зарегистрированы соответствующим уполномоченным федеральным органом исполнительной власти.
2. Государственной регистрации подлежат:
1) оригинальные лекарственные препараты;
2) воспроизведенные лекарственные препараты;
3) новые комбинации зарегистрированных ранее лекарственных препаратов;
4) лекарственные препараты, зарегистрированные ранее, но произведенные в других лекарственных формах, в новой дозировке.
3. Государственная регистрация лекарственных препаратов для медицинского применения осуществляется по результатам экспертизы лекарственных средств и этической экспертизы возможности проведения клинического исследования лекарственного препарата для медицинского применения (далее - этическая экспертиза). Государственная регистрация лекарственных препаратов для ветеринарного применения осуществляется по результатам экспертизы лекарственных средств для ветеринарного применения.
4. Государственная регистрация лекарственных препаратов осуществляется соответствующим уполномоченным федеральным органом исполнительной власти в срок, не превышающий двухсот десяти рабочих дней со дня принятия заявления о государственной регистрации лекарственного препарата. В указанный срок включается время, необходимое для повторного проведения экспертизы лекарственных средств и (или) этической экспертизы в соответствии со статьей 25 настоящего Федерального закона. Срок государственной регистрации лекарственного препарата исчисляется со дня принятия соответствующим уполномоченным федеральным органом исполнительной власти заявления о государственной регистрации лекарственного препарата с приложением необходимых документов по день выдачи регистрационного удостоверения лекарственного препарата. Время проведения клинического исследования лекарственного препарата, а также время, необходимое для направления уполномоченным федеральным органом исполнительной власти запроса о представлении необходимых материалов и представления заявителем ответа на данный запрос в соответствии со статьями 16, 19, 22 и 23 настоящего Федерального закона, не учитывается при исчислении срока государственной регистрации такого лекарственного препарата.
5. Государственной регистрации не подлежат:
1) лекарственные препараты, изготовленные аптечными организациями, ветеринарными аптечными организациями, индивидуальными предпринимателями, которые имеют лицензию на фармацевтическую деятельность, по рецептам на лекарственные препараты и требованиям медицинских организаций, ветеринарных организаций;
2) лекарственное растительное сырье;
3) лекарственные препараты, приобретенные физическими лицами за пределами территории Российской Федерации и предназначенные для личного использования;
4) лекарственные препараты, предназначенные для экспорта;
5) радиофармацевтические лекарственные препараты, изготовленные непосредственно в медицинских организациях в порядке, установленном уполномоченным федеральным органом исполнительной власти.
6. Не допускается государственная регистрация:
1) различных лекарственных препаратов под одинаковым торговым наименованием;
2) одного лекарственного препарата, выпускаемого производителем под различными торговыми наименованиями и представленного на государственную регистрацию в виде двух и более лекарственных препаратов.
Статья 14. Принципы экспертизы лекарственных средств и этической экспертизы
1. Экспертиза лекарственных средств и этическая экспертиза основываются на принципах законности, соблюдения прав и свобод человека и гражданина, прав юридического лица, независимости эксперта, объективности, всесторонности и полноты исследований, проводимых с использованием современных достижений науки и техники, ответственности федерального государственного бюджетного учреждения по проведению экспертизы лекарственных средств и экспертов за проведение и качество экспертизы.
2. Экспертиза лекарственных средств для медицинского применения проводится поэтапно:
1) на первом этапе - экспертиза документов для получения разрешения на проведение клинического исследования лекарственного препарата, за исключением:
а) лекарственных препаратов, которые разрешены для медицинского применения на территории Российской Федерации более двадцати лет и в отношении которых невозможно проведение исследования биоэквивалентности;
б) лекарственных препаратов для медицинского применения, в отношении которых проведены международные многоцентровые клинические исследования, часть из которых проведена на территории Российской Федерации;
2) на втором этапе - экспертиза предложенных методов контроля качества лекарственного средства и качества представленных образцов лекарственного средства с использованием этих методов (далее - экспертиза качества лекарственного средства) и экспертиза отношения ожидаемой пользы к возможному риску применения лекарственного препарата, осуществляемые после проведения его клинического исследования.
3. Экспертиза лекарственных средств для ветеринарного применения проводится одноэтапно и включает в себя экспертизу качества лекарственного средства и экспертизу отношения ожидаемой пользы к возможному риску применения лекарственного препарата.
Статья 15. Федеральное государственное бюджетное учреждение по проведению экспертизы лекарственных средств
Экспертиза лекарственных средств проводится федеральным государственным бюджетным учреждением соответствующего уполномоченного федерального органа исполнительной власти, созданным для обеспечения исполнения полномочий этого федерального органа по выдаче разрешений на проведение клинических исследований лекарственных препаратов и (или) по государственной регистрации лекарственных препаратов (далее - экспертное учреждение).
Статья 16. Организация проведения экспертизы лекарственных средств в целях их государственной регистрации
1. Экспертиза лекарственных средств проводится комиссией экспертов экспертного учреждения, назначенной его руководителем, на основании задания на проведение экспертизы лекарственного средства, выданного уполномоченным федеральным органом исполнительной власти. Руководитель экспертного учреждения обеспечивает надлежащее проведение экспертизы лекарственных средств в соответствии с заданием, выданным уполномоченным федеральным органом исполнительной власти, и организует подготовку сводного заключения этой комиссии. В состав этой комиссии по решению руководителя экспертного учреждения могут быть включены в качестве экспертов лица, не работающие в данном экспертном учреждении, если их специальные знания необходимы для проведения экспертизы и такие эксперты отсутствуют в данном экспертном учреждении.
2. Экспертом по проведению экспертизы лекарственных средств является аттестованный работник экспертного учреждения, который имеет высшее медицинское, фармацевтическое, биологическое, ветеринарное или химическое образование и проводит экспертизу лекарственных средств в порядке исполнения своих должностных обязанностей (далее - эксперт).
3. При проведении экспертизы лекарственных средств эксперт не может находиться в какой-либо зависимости от лица, назначившего эту экспертизу, разработчика лекарственного средства или других заинтересованных в результатах экспертизы лиц.
4. При проведении экспертизы лекарственных средств не допускается истребовать у заявителя либо иных лиц материалы, необходимые для проведения экспертизы. В случае недостаточности представленных эксперту материалов для дачи заключения эксперт ставит вопрос о представлении ему необходимых материалов перед руководителем экспертного учреждения, который обращается с соответствующим запросом в уполномоченный федеральный орган исполнительной власти, выдавший задание на проведение экспертизы лекарственного средства. Указанный федеральный орган исполнительной власти в течение пяти рабочих дней со дня поступления запроса руководителя экспертного учреждения направляет заявителю запрос о представлении необходимых материалов. Данный запрос может быть передан уполномоченному представителю заявителя лично под расписку, направлен по почте заказным письмом или передан в электронной форме по телекоммуникационным каналам связи. В случае направления данного запроса по почте заказным письмом он считается полученным по истечении шести дней с даты направления заказного письма.
4.1. Заявитель обязан представить ответ на запрос уполномоченного федерального органа исполнительной власти в срок, не превышающий девяноста рабочих дней со дня его получения. Уполномоченный федеральный орган исполнительной власти, выдавший задание на проведение экспертизы лекарственного средства, в течение пяти рабочих дней со дня поступления от заявителя ответа на данный запрос направляет такой ответ в экспертное учреждение. В случае непредставления по истечении девяноста рабочих дней заявителем ответа на данный запрос уполномоченный федеральный орган исполнительной власти, выдавший задание на проведение экспертизы лекарственного средства, в течение пяти рабочих дней направляет в экспертное учреждение уведомление о непредставлении заявителем ответа на запрос указанного органа. Время со дня направления запроса экспертного учреждения в уполномоченный федеральный орган исполнительной власти до дня получения экспертным учреждением ответа на запрос или уведомления о непредставлении ответа на запрос не учитывается при исчислении срока проведения экспертизы лекарственного средства.
5. Эксперт при проведении порученной ему руководителем экспертного учреждения экспертизы лекарственного средства обязан:
1) провести полное исследование представленных ему объектов, материалов, дать обоснованное и объективное заключение по поставленным перед ним вопросам или мотивированное заключение о невозможности проведения им экспертизы лекарственного средства, если поставленные вопросы выходят за пределы специальных знаний эксперта, объекты исследований и материалы непригодны или недостаточны для проведения исследований и дачи заключения либо современный уровень развития науки не позволяет ответить на поставленные вопросы;
2) не разглашать сведения, которые стали ему известны в связи с проведением экспертизы лекарственного средства, а также сведения, составляющие государственную, коммерческую или иную охраняемую законом тайну;
3) обеспечить сохранность представленных объектов исследований и материалов.
6. Эксперт не вправе:
1) проводить экспертизу лекарственного средства по обращению непосредственно к нему организаций или физических лиц;
2) самостоятельно собирать материалы для проведения экспертизы лекарственного средства;
3) проводить экспертизу лекарственного средства в качестве негосударственного эксперта.
7. В случае необходимости эксперт вправе ходатайствовать перед руководителем экспертного учреждения о привлечении к проведению экспертизы лекарственного средства других экспертов.
8. Каждый эксперт, входящий в состав комиссии экспертов, которой поручено проведение экспертизы лекарственного средства, независимо и самостоятельно проводит исследования, оценивает результаты, полученные им лично и другими экспертами, и формулирует выводы относительно поставленных вопросов в пределах своих специальных знаний.
9. Результаты экспертизы лекарственного средства оформляются заключением комиссии экспертов. В заключении комиссии экспертов указываются перечень исследований, объем проведенных каждым экспертом исследований, установленные каждым из них факты и сделанные в результате исследований выводы. Эксперт, мнение которого не совпадает с решением комиссии экспертов, вправе выразить свое мнение в письменной форме, которое приобщается к заключению комиссии экспертов.
10. Эксперты, входящие в состав комиссии, предупреждаются об ответственности в соответствии с законодательством Российской Федерации за дачу заключения, содержащего необоснованные или фальсифицированные выводы, о чем они дают подписку.
11. Определение уровня профессиональной подготовки экспертов и аттестация их на право проведения экспертизы лекарственных средств осуществляются экспертно-квалификационными комиссиями в порядке, установленном соответствующим уполномоченным федеральным органом исполнительной власти. Уровень профессиональной подготовки экспертов подлежит пересмотру указанными комиссиями не реже чем один раз в пять лет.
12. Правила проведения экспертизы лекарственных средств и форма заключения комиссии экспертов устанавливаются соответствующим уполномоченным федеральным органом исполнительной власти.
Статья 17. Этическая экспертиза
1. Этическая экспертиза проводится в целях выдачи заключения об этической обоснованности возможности проведения клинического исследования лекарственного препарата для медицинского применения советом по этике, созданным в порядке, установленном уполномоченным федеральным органом исполнительной власти.
2. Экспертами совета по этике могут быть представители медицинских, научных организаций, образовательных организаций высшего образования, а также представители общественных организаций, религиозных организаций и средств массовой информации. Данные эксперты не должны находиться в какой-либо зависимости от разработчиков лекарственных препаратов и других лиц, заинтересованных в результатах этической экспертизы.
3. Оплата услуг экспертов совета по этике осуществляется на основании договора, заключенного между уполномоченным федеральным органом исполнительной власти, которым создан совет по этике, и экспертом совета по этике, за счет бюджетных ассигнований, предусмотренных уполномоченному федеральному органу исполнительной власти, создавшему совет по этике, в федеральном бюджете на соответствующий год на обеспечение его деятельности, и в размерах, установленных Правительством Российской Федерации.
4. Эксперты совета по этике несут ответственность в соответствии с законодательством Российской Федерации.
5. Состав совета по этике, положение об этом совете, порядок его деятельности, требования к квалификации и опыту работы по экспертной оценке научных, медицинских и этических аспектов клинических исследований лекарственных препаратов для медицинского применения, предъявляемые к экспертам совета по этике, порядок организации и проведения этической экспертизы, форма заключения совета по этике устанавливаются уполномоченным федеральным органом исполнительной власти. Число представителей медицинских организаций не может превышать половину от общего числа экспертов совета по этике.
6. Информация о составе совета по этике, планах его работы и текущей деятельности размещается на официальном сайте уполномоченного федерального органа исполнительной власти в сети "Интернет" в установленном им порядке.
Статья 18. Подача и рассмотрение заявлений о государственной регистрации лекарственных препаратов и представление необходимых документов
1. Для государственной регистрации лекарственного препарата разработчик лекарственного препарата или уполномоченное им другое юридическое лицо (далее - заявитель) представляет в соответствующий уполномоченный федеральный орган исполнительной власти, осуществляющий государственную регистрацию лекарственных препаратов, заявление о государственной регистрации лекарственного препарата, а также в порядке, установленном соответствующим уполномоченным федеральным органом исполнительной власти, необходимые документы, из которых формируется регистрационное досье на лекарственный препарат (далее - регистрационное досье).
2. В заявлении о государственной регистрации лекарственного препарата указываются:
1) наименование и адрес заявителя и (или) производителя лекарственного препарата и адрес места осуществления производства лекарственного препарата;
2) наименование лекарственного препарата (международное непатентованное или химическое и торговое наименования);
3) перечень веществ, входящих в состав лекарственного препарата, с указанием количества каждого из них;
4) лекарственная форма, дозировка, способы введения и применения, срок годности лекарственного препарата;
5) описание фармакологических и фармакодинамических или иммунобиологических свойств лекарственного препарата;
6) заявленная производителем лекарственного препарата предельная отпускная цена на лекарственный препарат, включенный в перечень жизненно необходимых и важнейших лекарственных препаратов, в случае его государственной регистрации;
7) отсутствие необходимости проведения клинического исследования, исследования биоэквивалентности лекарственного препарата, разрешенного для медицинского применения на территории Российской Федерации более двадцати лет, с указанием нормативных правовых актов, подтверждающих данный срок применения.
3. Регистрационное досье формируется из следующих документов:
1) проекты макетов первичной упаковки и вторичной (потребительской) упаковки лекарственного препарата;
2) документ, переведенный на русский язык, подтверждающий соответствие производителя регистрируемого лекарственного препарата требованиям правил организации производства и контроля качества лекарственных средств, выданный компетентным органом страны производителя регистрируемого лекарственного препарата и заверенный в установленном порядке;
3) проект нормативной документации или нормативного документа на лекарственный препарат либо указание соответствующей фармакопейной статьи;
4) схема технологического процесса производства лекарственного препарата, ее описание и (или) схема технологического процесса производства фармацевтической субстанции, ее описание;
5) документ, переведенный на русский язык, подтверждающий соответствие производителя фармацевтической субстанции требованиям правил организации производства и контроля качества лекарственных средств, выданный компетентным органом страны производителя фармацевтической субстанции, заверенный в установленном порядке и содержащий:
а) наименование фармацевтической субстанции (международное непатентованное или химическое и торговое наименования);
б) наименование и адрес производителя фармацевтической субстанции;
в) срок годности фармацевтической субстанции;
6) документ, содержащий сведения о показателях качества фармацевтической субстанции, используемой при производстве лекарственных препаратов;
7) нормативная документация или нормативный документ на фармацевтическую субстанцию либо указание соответствующей фармакопейной статьи;
8) информация об условиях хранения, перевозки лекарственного препарата и иная информация;
9) отчет о результатах доклинического исследования лекарственного средства для медицинского применения, содержащий описание, результаты и статистический анализ результатов данного доклинического исследования;
10) отчет о результатах доклинического исследования лекарственного средства и клинического исследования лекарственного препарата для ветеринарного применения;
11) проект протокола клинического исследования лекарственного препарата для медицинского применения;
12) брошюра исследователя;
13) информационный листок пациента;
14) информация о выплатах и компенсациях пациентам (здоровым добровольцам, больным) (далее - пациенты), привлеченным к проведению клинических исследований лекарственного препарата для медицинского применения, исследований биоэквивалентности и (или) терапевтической эквивалентности;
15) отчет о результатах международных многоцентровых клинических исследований лекарственного препарата для медицинского применения, часть из которых проведена на территории Российской Федерации;
16) проект инструкции по применению лекарственного препарата, содержащий следующие сведения:
а) наименование лекарственного средства (международное непатентованное или химическое и торговое наименования);
б) лекарственная форма с указанием наименований и количественного содержания (активности) фармацевтических субстанций и вспомогательных веществ;
в) фармакотерапевтическая группа лекарственного препарата;
г) показания для применения;
д) противопоказания для применения;
е) режим дозирования, способ введения, при необходимости время приема лекарственного препарата, продолжительность лечения (в том числе у детей до и после одного года);
ж) меры предосторожности при применении;
з) симптомы передозировки, меры по оказанию помощи при передозировке;
и) указание, при необходимости, особенностей действия лекарственного препарата при первом приеме или при его отмене;
к) описание, при необходимости, действий врача (фельдшера), специалиста в области ветеринарии, пациента, владельца животного при пропуске приема одной или нескольких доз лекарственного препарата;
л) возможные побочные действия при применении лекарственного препарата;
м) взаимодействие с другими лекарственными препаратами и (или) пищевыми продуктами, кормами;
н) указание возможности и особенностей медицинского применения лекарственного препарата беременными женщинами, женщинами в период грудного вскармливания, детьми, взрослыми, имеющими хронические заболевания;
о) сведения о возможном влиянии лекарственного препарата для медицинского применения на способность управлять транспортными средствами, механизмами;
п) срок годности и указание на запрет применения лекарственного препарата по истечении срока годности;
р) условия хранения;
с) указание на необходимость хранения лекарственного препарата в местах, недоступных для детей;
т) указание, при необходимости, специальных мер предосторожности при уничтожении неиспользованных лекарственных препаратов;
у) сроки возможного использования продукции животного происхождения после введения животному лекарственного препарата для ветеринарного применения;
ф) наименование, адрес производителя лекарственного препарата и адрес места производства лекарственного препарата;
х) условия отпуска;
17) переведенная на русский язык и заверенная в установленном порядке копия документа, подтверждающего регистрацию лекарственного препарата в случае его регистрации вне пределов Российской Федерации;
18) документы, представляемые в соответствии со статьями 19 - 23 настоящего Федерального закона.
4. По желанию заявителя могут быть представлены отчеты о проведенных в стране заявителя и других странах результатах клинических исследований, исследований биоэквивалентности и (или) терапевтической эквивалентности лекарственного препарата (включая эпидемиологические или эпизоотологические исследования иммунобиологических лекарственных препаратов, предназначенных для иммунологической профилактики и лечения инфекционных заболеваний, в том числе у детей), содержащие описания проведенных исследований лекарственного препарата, их результаты и статистический анализ полученных результатов.
5. К заявлению о государственной регистрации лекарственного препарата прилагаются:
1) документ, подтверждающий уплату государственной пошлины за проведение экспертизы документов для получения разрешений на проведение клинических исследований лекарственного препарата для медицинского применения и этической экспертизы при обращении за государственной регистрацией лекарственного препарата;
2) документ, подтверждающий уплату государственной пошлины за проведение экспертизы качества лекарственного средства и экспертизы отношения ожидаемой пользы к возможному риску применения лекарственного препарата, разрешенного для медицинского применения на территории Российской Федерации более двадцати лет, при государственной регистрации лекарственного препарата;
3) документ, подтверждающий уплату государственной пошлины за проведение экспертизы качества лекарственного средства и экспертизы отношения ожидаемой пользы к возможному риску применения лекарственного препарата для медицинского применения, в отношении которого проведены международные многоцентровые клинические исследования, часть из которых проведена на территории Российской Федерации, при государственной регистрации лекарственного препарата;
4) документ, подтверждающий уплату государственной пошлины за проведение экспертизы качества лекарственного средства и экспертизы отношения ожидаемой пользы к возможному риску применения лекарственного препарата для ветеринарного применения при его государственной регистрации.
6. Не допускаются получение, разглашение, использование в коммерческих целях и в целях государственной регистрации лекарственных препаратов информации о результатах доклинических исследований лекарственных средств и клинических исследований лекарственных препаратов, представленной заявителем для государственной регистрации лекарственных препаратов, без его согласия в течение шести лет с даты государственной регистрации лекарственного препарата.
Несоблюдение запрета, установленного настоящей частью, влечет за собой ответственность в соответствии с законодательством Российской Федерации.
На территории Российской Федерации запрещается обращение лекарственных средств, зарегистрированных с нарушением настоящей части.
Статья 19. Принятие решения о выдаче экспертному учреждению и совету по этике задания на проведение экспертизы лекарственных средств
1. В течение пяти рабочих дней со дня принятия заявления о государственной регистрации лекарственного препарата уполномоченный федеральный орган исполнительной власти проводит проверку полноты и достоверности сведений, содержащихся в представленных заявителем материалах, и принимает решение о выдаче задания на проведение:
1) экспертизы лекарственных средств в части экспертизы документов для получения разрешения на проведение клинического исследования лекарственного препарата для медицинского применения в соответствии с целями, указанными в статье 38 настоящего Федерального закона, и этической экспертизы в отношении лекарственных препаратов, для которых не проводились клинические исследования на территории Российской Федерации, на основании документов, указанных в пунктах 1 - 9, 11 - 14, 17 части 3 и пункте 1 части 5 статьи 18 настоящего Федерального закона;
2) экспертизы лекарственных средств в части экспертизы качества лекарственного средства и экспертизы отношения ожидаемой пользы к возможному риску применения лекарственного препарата для медицинского применения в отношении лекарственных препаратов, разрешенных для медицинского применения на территории Российской Федерации более двадцати лет, на основании документов, указанных в пунктах 1 - 9, 16 части 3 и пункте 2 части 5 статьи 18 настоящего Федерального закона, а также лекарственных препаратов, в отношении которых проведены международные многоцентровые клинические исследования, часть из которых проведена на территории Российской Федерации, на основании документов, указанных в пунктах 1 - 9, 15 - 17 части 3 и пункте 3 части 5 статьи 18 настоящего Федерального закона;
3) экспертизы лекарственного средства в отношении лекарственных препаратов для ветеринарного применения на основании документов, указанных в пунктах 1 - 8, 10, подпунктах "а" - "д", "ж" - "м", "п" - "ф" пункта 16, пункте 17 части 3 и пункте 4 части 5 статьи 18 настоящего Федерального закона.
2. Уполномоченный федеральный орган исполнительной власти уведомляет в письменной форме заявителя о принятом решении или в случае отказа с указанием причин такого отказа.
2.1. В случае выявления недостоверности сведений, содержащихся в представленных заявителем материалах, уполномоченный федеральный орган исполнительной власти направляет заявителю запрос об уточнении указанных сведений. Данный запрос может быть передан уполномоченному представителю заявителя лично под расписку, направлен по почте заказным письмом или передан в электронной форме по телекоммуникационным каналам связи. В случае направления данного запроса по почте заказным письмом он считается полученным по истечении шести дней с даты направления заказного письма.
2.2. Заявитель обязан представить ответ на запрос уполномоченного федерального органа исполнительной власти в срок, не превышающий девяноста рабочих дней со дня получения данного запроса. Срок, указанный в части 1 настоящей статьи, приостанавливается со дня направления заявителю запроса уполномоченного федерального органа исполнительной власти до дня получения им ответа на данный запрос.
3. Основанием для отказа в организации экспертиз, указанных в части 1 настоящей статьи, является представление необходимых для проведения этих экспертиз документов, перечисленных в частях 3 и 5 статьи 18 настоящего Федерального закона, в неполном объеме, непредставление заявителем в установленный срок ответа на указанный в части 2.1 настоящей статьи запрос уполномоченного федерального органа исполнительной власти, а также представление документов, не содержащих исчерпывающего перечня необходимых сведений.
Статья 20. Экспертиза документов для получения разрешения на проведение клинического исследования лекарственного препарата для медицинского применения и этической экспертизы
1. Экспертиза документов для получения разрешения на проведение клинического исследования лекарственного препарата для медицинского применения в соответствии с целями, указанными в статье 38 настоящего Федерального закона, этическая экспертиза, составление комиссией экспертов и советом по этике заключений о возможности или невозможности проведения такого клинического исследования и направление этих заключений в уполномоченный федеральный орган исполнительной власти осуществляются в срок, не превышающий тридцати рабочих дней со дня получения экспертным учреждением задания уполномоченного федерального органа исполнительной власти с приложением необходимых документов, указанных в пунктах 9, 11, 12 части 3 статьи 18 настоящего Федерального закона, документов, указанных в части 4 статьи 18 настоящего Федерального закона и представленных по желанию заявителя, и советом по этике задания уполномоченного федерального органа исполнительной власти с приложением необходимых документов, указанных в пунктах 11 - 14 части 3 статьи 18 настоящего Федерального закона.
2. Документы, содержащиеся в регистрационном досье и поступившие в экспертное учреждение, совет по этике для осуществления их экспертизы в целях получения разрешения на проведение клинического исследования лекарственного препарата для медицинского применения, подлежат возврату в уполномоченный федеральный орган исполнительной власти одновременно с заключениями соответствующих экспертиз.
Статья 21. Получение разрешения на проведение клинического исследования лекарственного препарата для медицинского применения
1. В срок, не превышающий пяти рабочих дней со дня получения заключений, указанных в статье 20 настоящего Федерального закона, уполномоченный федеральный орган исполнительной власти осуществляет оценку поступивших заключений для определения их соответствия заданиям на проведение соответствующих экспертиз и уведомляет заявителя в письменной форме о результатах проведенных экспертиз и о возможности или невозможности выдачи заявителю разрешения на проведение клинического исследования лекарственного препарата для медицинского применения.
2. При принятии решения о возможности выдачи разрешения на проведение клинического исследования лекарственного препарата для медицинского применения уполномоченный федеральный орган исполнительной власти приостанавливает проведение государственной регистрации лекарственного препарата до дня подачи заявителем в уполномоченный федеральный орган исполнительной власти заявления о получении разрешения на проведение клинического исследования лекарственного препарата для медицинского применения.
3. В случае принятия решения о невозможности выдачи разрешения на проведение клинического исследования лекарственного препарата для медицинского применения уполномоченный федеральный орган исполнительной власти прекращает процедуру государственной регистрации лекарственного препарата.
Статья 22. Решение о проведении клинического исследования лекарственного препарата для медицинского применения
1. Для получения разрешения на проведение клинического исследования лекарственного препарата для медицинского применения заявитель представляет в уполномоченный федеральный орган исполнительной власти:
1) заявление о получении разрешения на проведение данного клинического исследования;
2) сведения об опыте работы исследователей по соответствующим специальностям и их опыте работы по проведению клинических исследований;
3) копию договора обязательного страхования жизни, здоровья пациентов, участвующих в клиническом исследовании лекарственного препарата для медицинского применения (далее - договор обязательного страхования), заключенного в соответствии с типовыми правилами обязательного страхования жизни, здоровья пациента, участвующего в клиническом исследовании лекарственного препарата для медицинского применения, утвержденными Правительством Российской Федерации (далее - типовые правила обязательного страхования), с указанием предельной численности пациентов, участвующих в клиническом исследовании лекарственного препарата для медицинского применения;
4) сведения о медицинских организациях, в которых предполагается проведение клинических исследований лекарственного препарата для медицинского применения (полное и сокращенное наименования, организационно-правовая форма, место нахождения и место осуществления деятельности, телефон, телефакс, адрес электронной почты медицинской организации);
5) предполагаемые сроки проведения клинического исследования лекарственного препарата для медицинского применения.
2. В срок, не превышающий пяти рабочих дней со дня принятия указанного в части 1 настоящей статьи заявления с приложением необходимых документов, уполномоченный федеральный орган исполнительной власти:
1) проводит проверку полноты и достоверности сведений, содержащихся в представленных заявителем материалах;
2) принимает решение о выдаче разрешения на проведение клинического исследования лекарственного препарата для медицинского применения или об отказе в выдаче указанного разрешения;
3) уведомляет в письменной форме заявителя о принятом решении или в случае отказа с указанием причин такого отказа;
4) выдает разрешение на проведение клинического исследования лекарственного препарата для медицинского применения в порядке, установленном уполномоченным федеральным органом исполнительной власти.
2.1. В случае выявления недостоверности сведений, содержащихся в представленных заявителем материалах, уполномоченный федеральный орган исполнительной власти направляет заявителю запрос об уточнении указанных сведений. Данный запрос может быть передан уполномоченному представителю заявителя лично под расписку, направлен по почте заказным письмом или передан в электронной форме по телекоммуникационным каналам связи. В случае направления данного запроса по почте заказным письмом он считается полученным по истечении шести дней с даты направления заказного письма.
2.2. Заявитель обязан представить ответ на запрос уполномоченного федерального органа исполнительной власти в срок, не превышающий девяноста рабочих дней со дня получения данного запроса. Срок, указанный в части 2 настоящей статьи, приостанавливается со дня направления заявителю запроса уполномоченного федерального органа исполнительной власти до дня получения им ответа на данный запрос.
3. Основаниями для отказа в выдаче разрешения на проведение клинического исследования лекарственного препарата для медицинского применения являются непредставление документов, указанных в части 1 настоящей статьи, несоответствие содержания представленных документов требованиям настоящего Федерального закона, непредставление заявителем в установленный срок ответа на указанный в части 2.1 настоящей статьи запрос уполномоченного федерального органа исполнительной власти либо наличие заключения комиссии экспертов или заключения совета по этике о невозможности проведения клинического исследования лекарственного препарата для медицинского применения по результатам проведенных экспертиз, предусмотренных статьей 20 настоящего Федерального закона.
Статья 23. Экспертиза качества лекарственного средства и экспертиза отношения ожидаемой пользы к возможному риску применения лекарственного препарата для медицинского применения
1. Экспертиза качества лекарственного средства и экспертиза отношения ожидаемой пользы к возможному риску применения лекарственного препарата для медицинского применения, составление комиссиями экспертов заключений по результатам проведенных экспертиз и направление таких заключений в уполномоченный федеральный орган исполнительной власти осуществляются в срок, не превышающий ста десяти рабочих дней со дня получения экспертным учреждением соответствующего задания уполномоченного федерального органа исполнительной власти с документами, указанными в пунктах 1 - 8, 15 - 17 части 3 статьи 18 настоящего Федерального закона, и отчета о проведенном клиническом исследовании лекарственного препарата для медицинского применения.
2. Для проведения указанных в части 1 настоящей статьи экспертиз заявитель представляет в уполномоченный федеральный орган исполнительной власти:
1) заявление о возобновлении государственной регистрации лекарственного препарата и проведении указанных в части 1 настоящей статьи экспертиз;
2) отчет о проведенном клиническом исследовании лекарственного препарата для медицинского применения;
3) документ, подтверждающий уплату государственной пошлины за проведение экспертизы качества лекарственного средства и экспертизы отношения ожидаемой пользы к возможному риску применения лекарственного препарата для медицинского применения при его государственной регистрации.
3. В срок, не превышающий пяти рабочих дней со дня принятия заявления с документами, указанными в части 1 и пунктах 2 и 3 части 2 настоящей статьи, уполномоченный федеральный орган исполнительной власти:
1) проводит проверку полноты и достоверности данных, содержащихся в представленном заявителем отчете о проведении клинического исследования лекарственного препарата для медицинского применения и в документах, содержащихся в регистрационном досье;
2) принимает решение о возобновлении государственной регистрации лекарственного препарата и проведении указанных в части 1 настоящей статьи экспертиз или об отказе в возобновлении государственной регистрации лекарственного препарата для медицинского применения и проведении таких экспертиз;
3) уведомляет в письменной форме заявителя о принятом решении или в случае отказа с указанием причин такого отказа.
3.1. В случае выявления недостоверности сведений, содержащихся в представленных заявителем материалах, уполномоченный федеральный орган исполнительной власти направляет заявителю запрос об уточнении указанных сведений. Данный запрос может быть передан уполномоченному представителю заявителя лично под расписку, направлен по почте заказным письмом или передан в электронной форме по телекоммуникационным каналам связи. В случае направления данного запроса по почте заказным письмом он считается полученным по истечении шести дней с даты направления заказного письма.
3.2. Заявитель обязан представить ответ на запрос уполномоченного федерального органа исполнительной власти в срок, не превышающий девяноста рабочих дней со дня получения данного запроса. Срок, указанный в части 3 настоящей статьи, приостанавливается со дня направления заявителю запроса уполномоченного федерального органа исполнительной власти до дня получения им ответа на данный запрос.
4. Основанием для отказа в возобновлении государственной регистрации лекарственного препарата и проведении указанных в части 1 настоящей статьи экспертиз является представление документов, указанных в части 1 и пунктах 2 и 3 части 2 настоящей статьи, в неполном объеме, непредставление заявителем в установленный срок ответа на указанный в части 3.1 настоящей статьи запрос уполномоченного федерального органа исполнительной власти или представление документов, не содержащих исчерпывающего перечня необходимых сведений.
5. В течение пятнадцати рабочих дней со дня получения решения уполномоченного федерального органа исполнительной власти о возобновлении государственной регистрации лекарственного препарата и проведении указанных в части 1 настоящей статьи экспертиз заявитель представляет в экспертное учреждение для проведения экспертизы качества лекарственного средства образцы лекарственного препарата для медицинского применения, произведенного в соответствии с требованиями опытно-промышленного и (или) промышленного регламентов, утвержденных руководителем производителя лекарственных средств, а также в соответствующих случаях образец фармацевтической субстанции, тест-штамма микроорганизмов, культуры клеток, образцы веществ, применяемых для контроля качества лекарственного средства путем сравнения с ними исследуемого лекарственного средства, в количествах, необходимых для воспроизведения методов контроля качества.
6. При получении образцов лекарственного препарата и фармацевтической субстанции экспертное учреждение выдает заявителю документ, подтверждающий получение указанных образцов, и в срок, не превышающий трех рабочих дней, уведомляет в письменной форме об этом уполномоченный федеральный орган исполнительной власти.
7. Срок для представления заявителем образцов лекарственного препарата и фармацевтической субстанции и срок уведомления экспертным учреждением об этом в письменной форме уполномоченного федерального органа исполнительной власти, указанные в частях 5 и 6 настоящей статьи, не включаются в срок проведения указанных в части 1 настоящей статьи экспертиз.
8. Документы, поступившие в экспертное учреждение для проведения указанных в части 1 настоящей статьи экспертиз, подлежат возврату в уполномоченный федеральный орган исполнительной власти одновременно с заключениями по результатам указанных экспертиз.
Статья 24. Экспертиза качества лекарственного средства и экспертиза отношения ожидаемой пользы к возможному риску применения лекарственного препарата для ветеринарного применения
1. Экспертиза качества лекарственного средства и экспертиза отношения ожидаемой пользы к возможному риску применения лекарственного препарата для ветеринарного применения, составление комиссиями экспертов заключений по результатам указанных экспертиз и направление таких заключений в уполномоченный федеральный орган исполнительной власти осуществляются в срок, не превышающий ста десяти рабочих дней со дня получения экспертным учреждением задания уполномоченного федерального органа исполнительной власти с документами, указанными в пунктах 1 - 8, 10, подпунктах "а" - "д", "ж" - "м", "п" - "ф" пункта 16 и пункте 17 части 3 статьи 18 настоящего Федерального закона.
2. В течение пятнадцати рабочих дней со дня получения решения уполномоченного федерального органа исполнительной власти о проведении указанных в части 1 настоящей статьи экспертиз заявитель представляет в экспертное учреждение для проведения экспертизы качества лекарственного средства образцы лекарственного препарата для ветеринарного применения, произведенного в соответствии с требованиями технологических регламентов, утвержденных руководителем производителя лекарственных средств, а также образец фармацевтической субстанции в количествах, необходимых для воспроизведения методов контроля качества.
3. При получении образцов лекарственного препарата и фармацевтической субстанции экспертное учреждение выдает заявителю документ, подтверждающий получение указанных образцов, и в срок, не превышающий трех рабочих дней, уведомляет в письменной форме об этом уполномоченный федеральный орган исполнительной власти.
4. Срок для представления заявителем образцов лекарственного препарата и фармацевтической субстанции и срок уведомления экспертным учреждением об этом в письменной форме уполномоченного федерального органа исполнительной власти, указанные в частях 2 и 3 настоящей статьи, не включаются в срок проведения указанных в части 1 настоящей статьи экспертиз.
5. Документы, поступившие в экспертное учреждение для проведения указанных в части 1 настоящей статьи экспертиз, подлежат возврату в уполномоченный федеральный орган исполнительной власти одновременно с заключениями по результатам указанных экспертиз.
Статья 25. Повторное проведение экспертизы лекарственных средств и этической экспертизы.
1. В случаях недостаточной обоснованности или неполноты заключения комиссии экспертов или совета по этике, наличия в нем противоречивых данных, фальсификации выводов экспертизы лекарственного средства и (или) этической экспертизы, сокрытия от уполномоченного федерального органа исполнительной власти оснований для отвода эксперта вследствие его заинтересованности в результатах соответствующей экспертизы, наличия данных о прямом либо косвенном вмешательстве в процесс соответствующей экспертизы лиц, не участвующих в ее проведении, но оказавших влияние на процесс и результаты ее проведения, уполномоченным федеральным органом исполнительной власти назначается повторная экспертиза лекарственного средства и (или) этическая экспертиза.
2. Повторная экспертиза лекарственного средства проводится в срок, установленный уполномоченным федеральным органом исполнительной власти и не превышающий сорока рабочих дней со дня получения экспертным учреждением задания на проведение повторной экспертизы лекарственного средства, повторная этическая экспертиза - в срок, не превышающий пятнадцати рабочих дней со дня получения советом по этике задания на проведение повторной этической экспертизы.
3. Финансовое обеспечение выполнения задания на проведение повторной экспертизы лекарственного средства не осуществляется, и средства, перечисленные ранее на проведение такой экспертизы, подлежат возврату в федеральный бюджет.
Статья 26. Ускоренная процедура экспертизы лекарственных средств
1. Ускоренная процедура экспертизы лекарственных средств в целях государственной регистрации лекарственных препаратов применяется в отношении воспроизведенных лекарственных препаратов. При проведении такой процедуры представляются информация, полученная при проведении клинических исследований лекарственных препаратов и опубликованная в специализированных печатных изданиях, а также документы, содержащие результаты исследования биоэквивалентности и (или) терапевтической эквивалентности лекарственного препарата для медицинского применения или результаты исследования биоэквивалентности лекарственного препарата для ветеринарного применения.
2. Ускоренная процедура экспертизы лекарственных средств не применяется в отношении иммунобиологических лекарственных препаратов, препаратов инсулина и лекарственных препаратов, впервые регистрируемых в Российской Федерации.
3. Ускоренная процедура экспертизы лекарственных средств проводится по решению соответствующего уполномоченного федерального органа исполнительной власти в срок, не превышающий шестидесяти рабочих дней. При этом экспертиза документов, содержащихся в регистрационном досье, для получения разрешения на проведение клинического исследования лекарственного препарата для медицинского применения и этическая экспертиза проводятся в срок, не превышающий пятнадцати рабочих дней, экспертиза качества лекарственного средства и экспертиза отношения ожидаемой пользы к возможному риску применения лекарственного препарата - в срок, не превышающий сорока пяти рабочих дней.
4. Ускоренная процедура экспертизы лекарственных средств проводится в порядке, установленном статьями 17 - 20, 23 и 24 настоящего Федерального закона, и не означает снижения требований к безопасности, качеству и эффективности лекарственных препаратов.
Статья 27. Решение о государственной регистрации лекарственного препарата.
1. В срок, не превышающий пяти рабочих дней со дня получения заключений комиссии экспертов по результатам экспертизы качества лекарственного средства и экспертизы отношения ожидаемой пользы к возможному риску применения лекарственного препарата, соответствующий уполномоченный федеральный орган исполнительной власти:
1) осуществляет оценку таких заключений для определения их соответствия заданию на проведение указанных экспертиз;
2) принимает решение о государственной регистрации лекарственного препарата или об отказе в государственной регистрации лекарственного препарата;
3) вносит при принятии решения о государственной регистрации лекарственного препарата данные о зарегистрированном лекарственном препарате, в том числе о фармацевтической субстанции, входящей в состав лекарственного препарата, в государственный реестр лекарственных средств и выдает заявителю регистрационное удостоверение лекарственного препарата, форма которого утверждается уполномоченным федеральным органом исполнительной власти, согласованные нормативную документацию, нормативный документ, инструкцию по применению лекарственного препарата и макеты первичной упаковки и вторичной (потребительской) упаковки с указанием на них номера регистрационного удостоверения лекарственного препарата и даты его государственной регистрации или в случае принятия решения об отказе в государственной регистрации лекарственного препарата уведомляет в письменной форме заявителя об этом с указанием причин такого отказа.
2. Основанием для отказа в государственной регистрации лекарственного препарата является решение соответствующего уполномоченного федерального органа исполнительной власти о том, что качество и (или) эффективность регистрируемого лекарственного препарата не подтверждены полученными данными или что риск причинения вреда здоровью человека или животного вследствие приема лекарственного препарата превышает эффективность его применения.
3. При государственной регистрации лекарственного препарата, включенного в перечень жизненно необходимых и важнейших лекарственных препаратов, необходимые данные заносятся в государственный реестр предельных отпускных цен производителей на лекарственные препараты, включенные в указанный перечень.
Статья 28. Регистрационное удостоверение лекарственного препарата
1. Регистрационное удостоверение лекарственного препарата с указанием лекарственных форм и дозировок выдается бессрочно, за исключением регистрационного удостоверения лекарственного препарата, выдаваемого со сроком действия пять лет, на впервые регистрируемые в Российской Федерации лекарственные препараты.
2. По истечении указанного в части 1 настоящей статьи срока выдается бессрочное регистрационное удостоверение лекарственного препарата при условии подтверждения его государственной регистрации.
Статья 29. Подтверждение государственной регистрации лекарственного препарата
1. Подтверждение государственной регистрации лекарственного препарата осуществляется при выдаче бессрочного регистрационного удостоверения лекарственного препарата в случае, указанном в части 2 статьи 28 настоящего Федерального закона, в срок, не превышающий девяноста рабочих дней со дня получения соответствующим уполномоченным федеральным органом исполнительной власти заявления о подтверждении государственной регистрации лекарственного препарата, оформленного в соответствии с частью 2 статьи 18 настоящего Федерального закона.
2. Подтверждение государственной регистрации лекарственного препарата осуществляется по результатам экспертизы отношения ожидаемой пользы к возможному риску применения лекарственного препарата, а также экспертизы качества лекарственного средства, проводимой в случае внесения изменений в нормативную документацию или нормативный документ.
3. К заявлению о подтверждении государственной регистрации лекарственного препарата прилагаются документ, подтверждающий уплату государственной пошлины за подтверждение государственной регистрации лекарственного препарата для медицинского применения или лекарственного препарата для ветеринарного применения, документ, содержащий результаты мониторинга безопасности лекарственного препарата, проводимого заявителем, по форме, установленной соответствующим уполномоченным федеральным органом исполнительной власти. Нормативная документация или нормативный документ, проект инструкции по применению лекарственного препарата, проекты макетов первичной упаковки и вторичной (потребительской) упаковки лекарственного препарата прилагаются к заявлению о подтверждении государственной регистрации лекарственного препарата вновь только в случае, если в них вносятся изменения.
4. В течение десяти рабочих дней со дня принятия заявления о подтверждении государственной регистрации лекарственного препарата и необходимых документов соответствующий уполномоченный федеральный орган исполнительной власти:
1) проводит проверку полноты и достоверности сведений, содержащихся в представленных заявителем материалах;
2) принимает решение о проведении или об отказе в проведении экспертизы отношения ожидаемой пользы к возможному риску применения лекарственного препарата, а также экспертизы качества лекарственного средства, проводимой в случае внесения изменений в нормативную документацию или нормативный документ;
3) уведомляет в письменной форме заявителя о принятом решении или в случае принятия решения об отказе в проведении экспертизы с указанием причин такого отказа.
4.1. В случае выявления недостоверности сведений, содержащихся в представленных заявителем материалах, уполномоченный федеральный орган исполнительной власти направляет заявителю запрос об уточнении указанных сведений. Данный запрос может быть передан уполномоченному представителю заявителя лично под расписку, направлен по почте заказным письмом или передан в электронной форме по телекоммуникационным каналам связи. В случае направления данного запроса по почте заказным письмом он считается полученным по истечении шести дней с даты направления заказного письма.
4.2. Заявитель обязан представить ответ на запрос уполномоченного федерального органа исполнительной власти в срок, не превышающий девяноста рабочих дней со дня получения данного запроса. Срок, указанный в части 4 настоящей статьи, приостанавливается со дня направления заявителю запроса уполномоченного федерального органа исполнительной власти до дня получения им ответа на данный запрос и не учитывается при исчислении срока подтверждения государственной регистрации лекарственного препарата.
5. Основанием для отказа в проведении экспертизы отношения ожидаемой пользы к возможному риску применения лекарственного препарата и (или) экспертизы качества лекарственного средства является представление документов, указанных в частях 1 и 3 настоящей статьи, в неполном объеме, непредставление заявителем в установленный срок ответа на указанный в части 4.1 настоящей статьи запрос уполномоченного федерального органа исполнительной власти, а также отсутствие в представленных документах исчерпывающих сведений, которые должны быть отражены в них.
6. Экспертиза отношения ожидаемой пользы к возможному риску применения лекарственного препарата и (или) экспертиза качества лекарственного средства в целях подтверждения государственной регистрации лекарственного препарата проводятся на основании документов, указанных в части 3 настоящей статьи, в порядке, установленном частями 5 - 8 статьи 23 и статьей 24 настоящего Федерального закона.
7. В период проведения процедуры подтверждения государственной регистрации лекарственного препарата его гражданский оборот осуществляется на территории Российской Федерации.
Статья 30. Внесение изменений в документы, содержащиеся в регистрационном досье на зарегистрированный лекарственный препарат для медицинского применения
1. В целях внесения изменений в документы, содержащиеся в регистрационном досье на зарегистрированный лекарственный препарат для медицинского применения, заявитель представляет в уполномоченный федеральный орган исполнительной власти заявление о внесении таких изменений по форме, установленной уполномоченным федеральным органом исполнительной власти, и приложенные к нему изменения в указанные документы, а также документы, подтверждающие необходимость внесения таких изменений. Принятие решения о внесении таких изменений или об отказе в их внесении осуществляется в срок, не превышающий девяноста рабочих дней со дня принятия уполномоченным федеральным органом исполнительной власти заявления о внесении таких изменений.
2. В случае внесения изменений в инструкцию по применению лекарственного препарата в отношении сведений, указанных в подпунктах "г" - "п", "х" пункта 16 части 3 статьи 18 настоящего Федерального закона, в состав лекарственного препарата для медицинского применения, изменения места производства лекарственного препарата для медицинского применения, изменения показателей качества лекарственного препарата для медицинского применения и (или) методов контроля качества лекарственного препарата для медицинского применения, изменения срока годности лекарственного препарата для медицинского применения проводится экспертиза лекарственных средств в части экспертизы качества лекарственного средства и (или) экспертизы отношения ожидаемой пользы к возможному риску применения лекарственного препарата для медицинского применения. В случае необходимости внесения иных изменений в данную инструкцию экспертиза лекарственного средства для внесения таких изменений в сведения о зарегистрированном лекарственном препарате не проводится.
3. С заявлением о внесении изменений в документы, содержащиеся в регистрационном досье на зарегистрированный лекарственный препарат для медицинского применения, наряду с документами, указанными в части 1 настоящей статьи, представляются документы, подтверждающие уплату государственной пошлины за внесение изменений в инструкцию по применению лекарственного препарата для медицинского применения, государственной пошлины за внесение изменений в состав лекарственного препарата для медицинского применения.
4. В течение десяти рабочих дней со дня поступления указанного в части 1 настоящей статьи заявления и необходимых документов уполномоченный федеральный орган исполнительной власти:
1) проводит проверку полноты и достоверности сведений, содержащихся в представленных заявителем материалах;
2) принимает решение о проведении указанных в части 2 настоящей статьи соответствующих экспертиз лекарственного средства или об отказе в их проведении;
3) уведомляет в письменной форме заявителя о принятом решении или в случае принятия решения об отказе в проведении соответствующей экспертизы с указанием причин такого отказа.
4.1. В случае выявления недостоверности сведений, содержащихся в представленных заявителем материалах, уполномоченный федеральный орган исполнительной власти направляет заявителю запрос об уточнении указанных сведений. Данный запрос может быть передан уполномоченному представителю заявителя лично под расписку, направлен по почте заказным письмом или передан в электронной форме по телекоммуникационным каналам связи. В случае направления данного запроса по почте заказным письмом он считается полученным по истечении шести дней с даты направления заказного письма.
4.2. Заявитель обязан представить ответ на запрос уполномоченного федерального органа исполнительной власти в срок, не превышающий девяноста рабочих дней со дня получения данного запроса. Срок, указанный в части 4 настоящей статьи, приостанавливается со дня направления заявителю запроса уполномоченного федерального органа исполнительной власти до дня получения им ответа на данный запрос и не учитывается при исчислении срока принятия решения о внесении изменений в документы, содержащиеся в регистрационном досье на зарегистрированный лекарственный препарат для медицинского применения.
5. Основанием для отказа в проведении указанных в части 2 настоящей статьи экспертиз является представление документов, перечисленных в частях 1 и 3 настоящей статьи, в неполном объеме, непредставление заявителем в установленный срок ответа на указанный в части 4.1 настоящей статьи запрос уполномоченного федерального органа исполнительной власти, а также отсутствие в представленных документах достаточных сведений, подтверждающих необходимость внесения изменений.
6. Указанные в части 2 настоящей статьи экспертизы проводятся в порядке, установленном статьей 23 настоящего Федерального закона.
7. В срок, не превышающий пяти рабочих дней со дня получения заключений комиссий экспертов по результатам указанных в части 2 настоящей статьи экспертиз, уполномоченный федеральный орган исполнительной власти:
1) принимает решение о внесении изменений в документы, содержащиеся в регистрационном досье на зарегистрированный лекарственный препарат для медицинского применения, или об отказе во внесении таких изменений;
2) вносит в государственный реестр лекарственных средств на основании решения о внесении изменений в документы, содержащиеся в регистрационном досье на зарегистрированный лекарственный препарат для медицинского применения, необходимые изменения и возвращает их заявителю.
8. Основанием для отказа во внесении изменений в документы, содержащиеся в регистрационном досье на зарегистрированный лекарственный препарат для медицинского применения, является заключение уполномоченного федерального органа исполнительной власти о возможности снижения безопасности, качества, эффективности лекарственного средства в случае внесения таких изменений.
9. Допускается гражданский оборот лекарственных препаратов для медицинского применения, произведенных до принятия уполномоченным федеральным органом исполнительной власти решения о внесении изменений в документы, содержащиеся в регистрационном досье на такие лекарственные препараты, или решения об отказе во внесении указанных изменений.
Статья 31. Внесение изменений в документы, содержащиеся в регистрационном досье на зарегистрированный лекарственный препарат для ветеринарного применения
1. В целях внесения изменений в документы, содержащиеся в регистрационном досье на зарегистрированный лекарственный препарат для ветеринарного применения, заявитель представляет в уполномоченный федеральный орган исполнительной власти заявление о внесении таких изменений по форме, установленной уполномоченным федеральным органом исполнительной власти, и приложенные к нему изменения в указанные документы, а также документы, подтверждающие необходимость внесения таких изменений. Принятие решения о внесении таких изменений или об отказе в их внесении осуществляется в срок, не превышающий девяноста рабочих дней со дня принятия уполномоченным федеральным органом исполнительной власти заявления о внесении таких изменений.
2. В случае внесения изменений в инструкцию по применению лекарственного препарата для ветеринарного применения в отношении сведений об изменениях дозировки, сроков возможного использования продукции животного происхождения после применения лекарственного препарата для ветеринарного применения проводится экспертиза лекарственного средства для ветеринарного применения. В случае необходимости внесения иных изменений в данную инструкцию экспертиза лекарственного средства для ветеринарного применения не проводится.
3. С заявлением о внесении изменений в документы, содержащиеся в регистрационном досье на зарегистрированный лекарственный препарат для ветеринарного применения, наряду с документами, указанными в части 1 настоящей статьи, представляются документы, подтверждающие уплату государственной пошлины за внесение изменений в инструкцию по применению лекарственного препарата для ветеринарного применения.
4. В срок, не превышающий десяти рабочих дней со дня поступления указанного в части 1 настоящей статьи заявления и необходимых документов, уполномоченный федеральный орган исполнительной власти:
1) проводит проверку полноты и достоверности сведений, содержащихся в представленных заявителем документах;
2) принимает решение о проведении экспертизы лекарственного средства для ветеринарного применения или об отказе в ее проведении;
3) уведомляет в письменной форме заявителя о принятом решении или в случае принятия решения об отказе в проведении экспертизы лекарственного средства для ветеринарного применения с указанием причин такого отказа.
5. Основанием для отказа в проведении экспертизы лекарственного средства для ветеринарного применения является представление документов, указанных в частях 1 и 3 настоящей статьи, в неполном объеме или отсутствие в представленных документах достаточных сведений, подтверждающих необходимость внесения изменений.
6. Экспертиза лекарственного средства для ветеринарного применения в целях внесения изменений в документы, содержащиеся в регистрационном досье на зарегистрированный лекарственный препарат для ветеринарного применения, осуществляется в порядке, установленном статьей 24 настоящего Федерального закона.
7. Основанием для отказа во внесении изменений в документы, содержащиеся в регистрационном досье на зарегистрированный лекарственный препарат для ветеринарного применения, является заключение уполномоченного федерального органа исполнительной власти о возможности снижения безопасности, качества, эффективности лекарственного препарата для ветеринарного применения в случае внесения изменений в указанные документы.
8. Допускается гражданский оборот лекарственных препаратов для ветеринарного применения, произведенных до принятия уполномоченным федеральным органом исполнительной власти решения о внесении изменений в документы, содержащиеся в регистрационном досье на зарегистрированный лекарственный препарат для ветеринарного применения, или об отказе во внесении указанных изменений.
Статья 32. Отмена государственной регистрации лекарственного препарата
Решение об отмене государственной регистрации лекарственного препарата и исключении лекарственного препарата из государственного реестра лекарственных средств принимается уполномоченным федеральным органом исполнительной власти в случае:
1) представления соответствующим уполномоченным федеральным органом исполнительной власти заключения о риске или об угрозе здоровью, жизни человека или животного при применении лекарственного препарата, превышающих его эффективность, по результатам осуществляемого им мониторинга безопасности лекарственного препарата;
2) подачи разработчиком лекарственного средства или уполномоченным им другим юридическим лицом заявления об отмене государственной регистрации лекарственного препарата;
3) неподтверждения государственной регистрации лекарственного препарата по истечении срока действия регистрационного удостоверения, выданного на пять лет;
4) непредставления заявителем информации, которая может повлечь за собой необходимость внесения изменений в документы, содержащиеся в регистрационном досье на зарегистрированный лекарственный препарат, в течение тридцати рабочих дней со дня наступления этих изменений;
5) осуществления государственной регистрации лекарственного препарата под торговым наименованием зарегистрированного ранее под этим торговым наименованием лекарственного препарата;
6) осуществления государственной регистрации заявителем одного и того же лекарственного препарата под различными торговыми наименованиями;
7) вынесения судом решения о нарушении прав правообладателя объектов интеллектуальной собственности при обращении лекарственных средств.
Статья 33. Государственный реестр лекарственных средств
1. Государственный реестр лекарственных средств содержит перечень лекарственных препаратов, прошедших государственную регистрацию, перечень фармацевтических субстанций, входящих в состав лекарственных препаратов, и следующую информацию:
1) в отношении лекарственных препаратов:
а) наименование лекарственного препарата (международное непатентованное или химическое и торговое наименования);
б) лекарственная форма с указанием дозировки лекарственного препарата и его количества в потребительской упаковке;
в) наименование разработчика лекарственного препарата;
г) наименование и адрес производителя лекарственного препарата;
д) фармакотерапевтическая группа лекарственного препарата;
е) показания и противопоказания к применению лекарственного препарата;
ж) побочные действия лекарственного препарата;
з) срок годности лекарственного препарата;
и) условия хранения лекарственного препарата;
к) условия отпуска лекарственного препарата;
л) номер фармакопейной статьи или в случае ее отсутствия номер нормативной документации либо нормативного документа;
м) дата государственной регистрации лекарственного препарата и его регистрационный номер;
2) в отношении фармацевтических субстанций:
а) наименование фармацевтической субстанции (международное непатентованное или химическое и торговое наименования);
б) наименование и адрес производителя фармацевтической субстанции;
в) срок годности фармацевтической субстанции;
г) условия хранения фармацевтической субстанции;
д) номер фармакопейной статьи или в случае ее отсутствия номер нормативной документации либо нормативного документа.
2. Фармацевтическая субстанция, неиспользуемая при производстве лекарственных препаратов, может быть включена в государственный реестр лекарственных средств на основании заявления разработчика, производителя лекарственного средства либо уполномоченного ими юридического лица при условии проведения в отношении такой фармацевтической субстанции экспертизы качества фармацевтической субстанции в порядке, установленном статьей 34 настоящего Федерального закона.
3. Порядок ведения государственного реестра лекарственных средств для медицинского применения и порядок ведения государственного реестра лекарственных средств для ветеринарного применения утверждаются соответствующим уполномоченным федеральным органом исполнительной власти.
Статья 34. Экспертиза качества фармацевтической субстанции, неиспользуемой при производстве лекарственных препаратов
1. Для включения фармацевтической субстанции, неиспользуемой при производстве лекарственных препаратов, в государственный реестр лекарственных средств, проводится экспертиза ее качества.
2. Экспертиза качества указанной в части 1 настоящей статьи фармацевтической субстанции, составление комиссией экспертов заключений по результатам такой экспертизы и их направление в уполномоченный федеральный орган исполнительной власти осуществляются в срок, не превышающий шестидесяти рабочих дней со дня получения экспертным учреждением соответствующего задания уполномоченного федерального органа исполнительной власти и документов, указанных в пунктах 4 - 7 части 3 статьи 18 настоящего Федерального закона.
3. Для проведения экспертизы качества указанной в части 1 настоящей статьи фармацевтической субстанции заявитель представляет в уполномоченный федеральный орган исполнительной власти:
1) заявление о включении в государственный реестр лекарственных средств данной фармацевтической субстанции;
2) документ, подтверждающий уплату государственной пошлины за включение фармацевтической субстанции, неиспользуемой при производстве лекарственных препаратов, в государственный реестр лекарственных средств;
3) документы, указанные в пунктах 4 - 7 части 3 статьи 18 настоящего Федерального закона.
4. В течение пяти рабочих дней со дня принятия заявления о включении указанной в части 1 настоящей статьи фармацевтической субстанции в государственный реестр лекарственных средств и документов, перечисленных в части 1 и пункте 2 части 3 настоящей статьи, уполномоченный федеральный орган исполнительной власти:
1) проводит проверку полноты и достоверности данных, содержащихся в представленных заявителем документах;
2) принимает решение о направлении в экспертное учреждение задания на проведение экспертизы качества указанной в части 1 настоящей статьи фармацевтической субстанции или об отказе в таком направлении;
3) уведомляет в письменной форме заявителя о принятом решении или в случае отказа с указанием причин такого отказа.
4.1. В случае выявления недостоверности сведений, содержащихся в представленных заявителем материалах, уполномоченный федеральный орган исполнительной власти направляет заявителю запрос об уточнении указанных сведений. Данный запрос может быть передан уполномоченному представителю заявителя лично под расписку, направлен по почте заказным письмом или передан в электронной форме по телекоммуникационным каналам связи. В случае направления данного запроса по почте заказным письмом он считается полученным по истечении шести дней с даты направления заказного письма.
4.2. Заявитель обязан представить ответ на запрос уполномоченного федерального органа исполнительной власти в срок, не превышающий девяноста рабочих дней со дня получения данного запроса. Срок, указанный в части 4 настоящей статьи, приостанавливается со дня направления заявителю запроса уполномоченного федерального органа исполнительной власти до дня получения им ответа на данный запрос и не учитывается при исчислении срока экспертизы качества фармацевтической субстанции, неиспользуемой при производстве лекарственных препаратов.
5. Основанием для отказа в направлении в экспертное учреждение задания на проведение экспертизы качества указанной в части 1 настоящей статьи фармацевтической субстанции является непредставление документов, перечисленных в части 2 и пункте 2 части 3 настоящей статьи, непредставление заявителем в установленный срок ответа на указанный в части 4.1 настоящей статьи запрос уполномоченного федерального органа исполнительной власти или представление документов, не содержащих исчерпывающего перечня необходимых сведений.
6. В течение пятнадцати рабочих дней со дня получения решения уполномоченного федерального органа исполнительной власти о направлении в экспертное учреждение задания на проведение экспертизы качества указанной в части 1 настоящей статьи фармацевтической субстанции заявитель представляет в экспертное учреждение для проведения данной экспертизы образцы фармацевтической субстанции в количестве, необходимом для воспроизведения методов контроля качества. Экспертное учреждение при получении образцов указанной в части 1 настоящей статьи фармацевтической субстанции выдает заявителю документ, подтверждающий получение этих образцов, и в срок, не превышающий трех рабочих дней, уведомляет в письменной форме об этом уполномоченный федеральный орган исполнительной власти. Эти сроки не включаются в срок проведения данной экспертизы.
7. Документы, поступившие в экспертное учреждение для проведения экспертизы качества указанной в части 1 настоящей статьи фармацевтической субстанции, подлежат возврату в уполномоченный федеральный орган исполнительной власти одновременно с заключениями по результатам проведенной экспертизы.
8. В срок, не превышающий пяти рабочих дней со дня получения заключения комиссии экспертов по результатам экспертизы качества указанной в части 1 настоящей статьи фармацевтической субстанции, уполномоченный федеральный орган исполнительной власти:
1) осуществляет оценку такого заключения для определения его соответствия заданию на проведение данной экспертизы;
2) принимает решение о включении указанной в части 1 настоящей статьи фармацевтической субстанции в государственный реестр лекарственных средств или решение об отказе в таком включении;
3) вносит при принятии решения о включении указанной в части 1 настоящей статьи фармацевтической субстанции в государственный реестр лекарственных средств предусмотренную пунктом 2 части 1 статьи 33 настоящего Федерального закона информацию и уведомляет об этом в письменной форме заявителя.
Статья 35. Повторное представление лекарственного препарата, не прошедшего государственной регистрации лекарственных препаратов, на государственную регистрацию лекарственных препаратов
Повторное представление в соответствующий уполномоченный федеральный орган исполнительной власти лекарственного препарата, не прошедшего государственной регистрации лекарственных препаратов или получившего отказ в указанной регистрации и впоследствии подвергшегося изменению в части его состава, рассматривается как представление нового лекарственного препарата на его государственную регистрацию независимо от сохранения его первичного наименования.
Статья 36. Обжалование решения об отказе в выдаче разрешения на проведение клинических исследований лекарственного препарата или решения об отказе в государственной регистрации лекарственного препарата
Решение соответствующего уполномоченного федерального органа исполнительной власти об отказе в выдаче разрешения на проведение клинических исследований лекарственного препарата или решение об отказе в государственной регистрации лекарственного препарата может быть обжаловано в порядке, установленном законодательством Российской Федерации.
Статья 37. Информация, связанная с осуществлением государственной регистрации лекарственных препаратов, информация о зарегистрированных лекарственных препаратах и лекарственных препаратах, исключенных из государственного реестра лекарственных средств
1. Соответствующий уполномоченный федеральный орган исполнительной власти размещает на своем официальном сайте в сети "Интернет" информацию, связанную с осуществлением государственной регистрации лекарственных препаратов, в том числе проведением экспертизы лекарственных средств, информацию о зарегистрированных лекарственных препаратах и лекарственных препаратах, исключенных из государственного реестра лекарственных средств, не позднее чем через пять рабочих дней со дня получения соответствующим уполномоченным федеральным органом исполнительной власти заявления о государственной регистрации лекарственного препарата.
2. Сроки и порядок размещения указанной в части 1 настоящей статьи информации устанавливаются уполномоченным федеральным органом исполнительной власти.
Скачано с www.znanio.ru

















































Материалы на данной страницы взяты из открытых источников либо размещены пользователем в соответствии с договором-офертой сайта. Вы можете сообщить о нарушении.