Поделиться


Синдром Альпорта
(Гематурический нефрит, Наследственный нефрит 1 типа, Семейный гломерулонефрит)
Общие сведения
Синдром Альпорта – наследственное заболевание почек, вызванное изменением синтеза коллагена типа IV, образующего базальные мембраны почечных клубочков, структуры внутреннего уха, хрусталика глаза.
Мужчины страдают развернутой формой болезни с тяжелой симптоматикой.
Женщины часто являются носителями гена, оставаясь здоровыми, или проявления болезни у них выражены слабо.
Общие сведения
Основные симптомы:
микрогематурия,
протеинурия,
почечная недостаточность,
сенсорная тугоухость,
деформация и вывих хрусталика,
катаракта.
Диагноз устанавливается согласно клинико-анамнестическим данным, результатам общего анализа мочи, исследования биоптата почки, аудиометрии и офтальмологического осмотра. Лечение симптоматическое, включает терапию иАПФ и БРА.

История
Артур Сесил Альпорт – английский врач.
В 1927 г. описал три поколения в семье, у которой прогрессирующий наследственный нефрит сочетался с тугоухостью. Кроме того заметил, что самым частым симптомом была гематурия, а мужчины поражались более тяжело, чем женщины. Впоследствии, было описано много подобных семейств, а в 1961 г. болезнь назвали синдромом Альпорта.

Распространенность
Редкое заболевание, но на его долю приходится более 1 % всех пациентов, получающих заместительную почечную терапию.
В литературе приводятся различные данные: от 1:5000 в штате Юта, США, до 1:53 000 новорожденных в Финляндии.
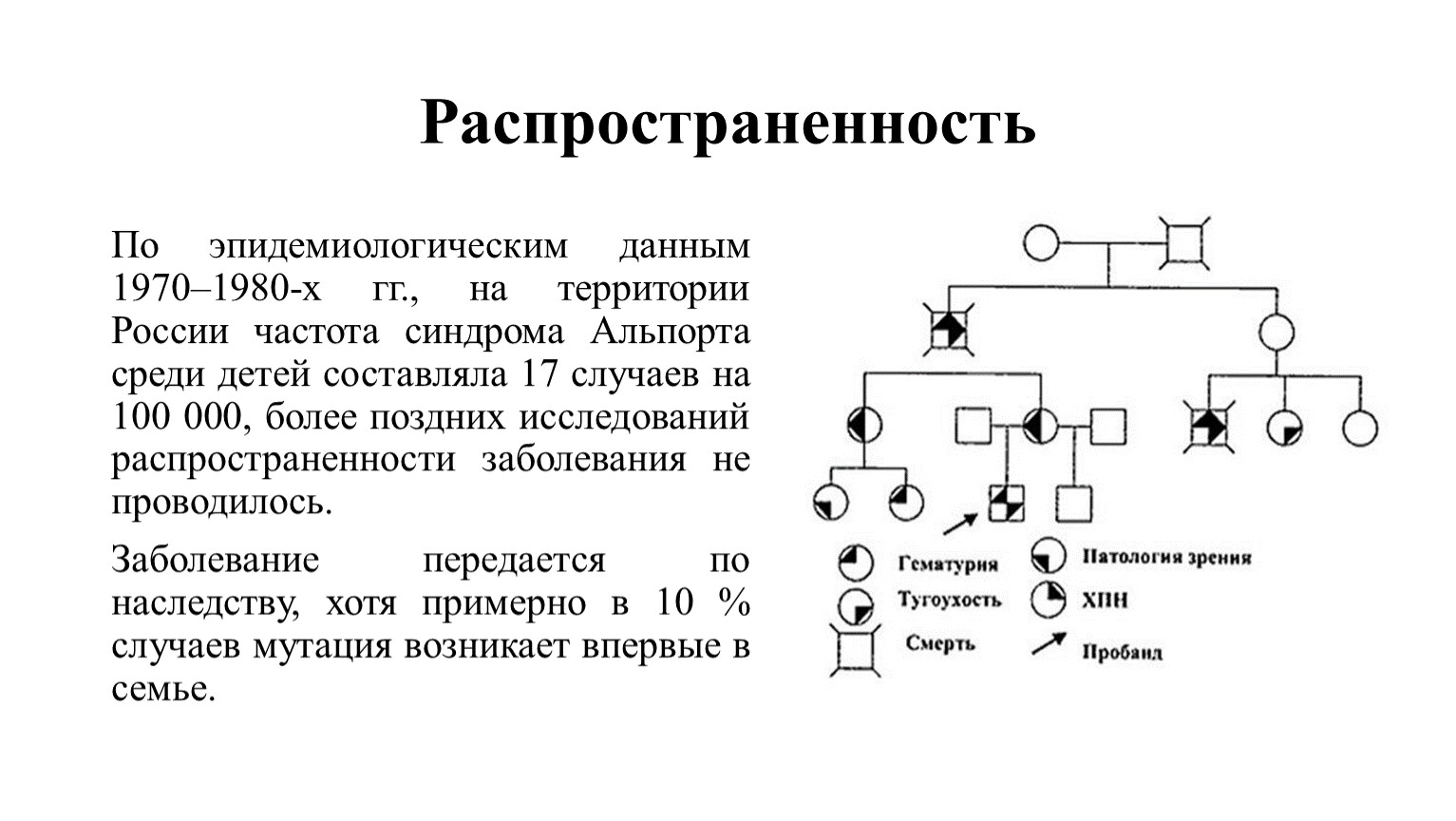
Распространенность
По эпидемиологическим данным 1970–1980-х гг., на территории России частота синдрома Альпорта среди детей составляла 17 случаев на 100 000, более поздних исследований распространенности заболевания не проводилось.
Заболевание передается по наследству, хотя примерно в 10 % случаев мутация возникает впервые в семье.

Причины
По своей природе синдром является гетерогенным наследственным заболеванием – его развитие провоцируется дефектом генов, которые кодируют структуру различных цепей IV типа коллагена.
Генетические изменения представлены:
делециями,
сплайсинг,
миссенс
нонсенс-мутациями.

Причины
Их локализация определяет тип наследования болезни:
X-сцепленный доминантный. Связан с мутацией в локусе COL4A5, который находится на половой хромосоме X. Ген кодирует а5-цепь коллагена 4 типа. Данный генетический дефект обуславливает 80-85% случаев наследственного нефрита. В полной мере заболевание проявляется у мальчиков и мужчин, у представительниц женского пола оставшийся нормальный ген в X-хромосоме компенсирует производство функционального коллагена.

Причины
Аутосомно-рецессивный. Развивается на основе мутаций в генах C0L4A3 и COL4A4. Они локализованы на второй хромосоме, отвечают за структуру а3- и а4-цепи коллагена. Пациенты с этим вариантом синдрома составляют около 15% больных. Выраженность симптомов не зависит от пола.
Аутосомно-доминантный. Нефрит возникает в результате мутаций генов COL4A3-COLA4, находящихся на 2 хромосоме. Как и в случае аутосомно-рецессивной формой болезни, нарушается синтез а4- и а3-цепей коллагена четвертого типа. Распространенность – 1% всех случаев генетического нефрита.
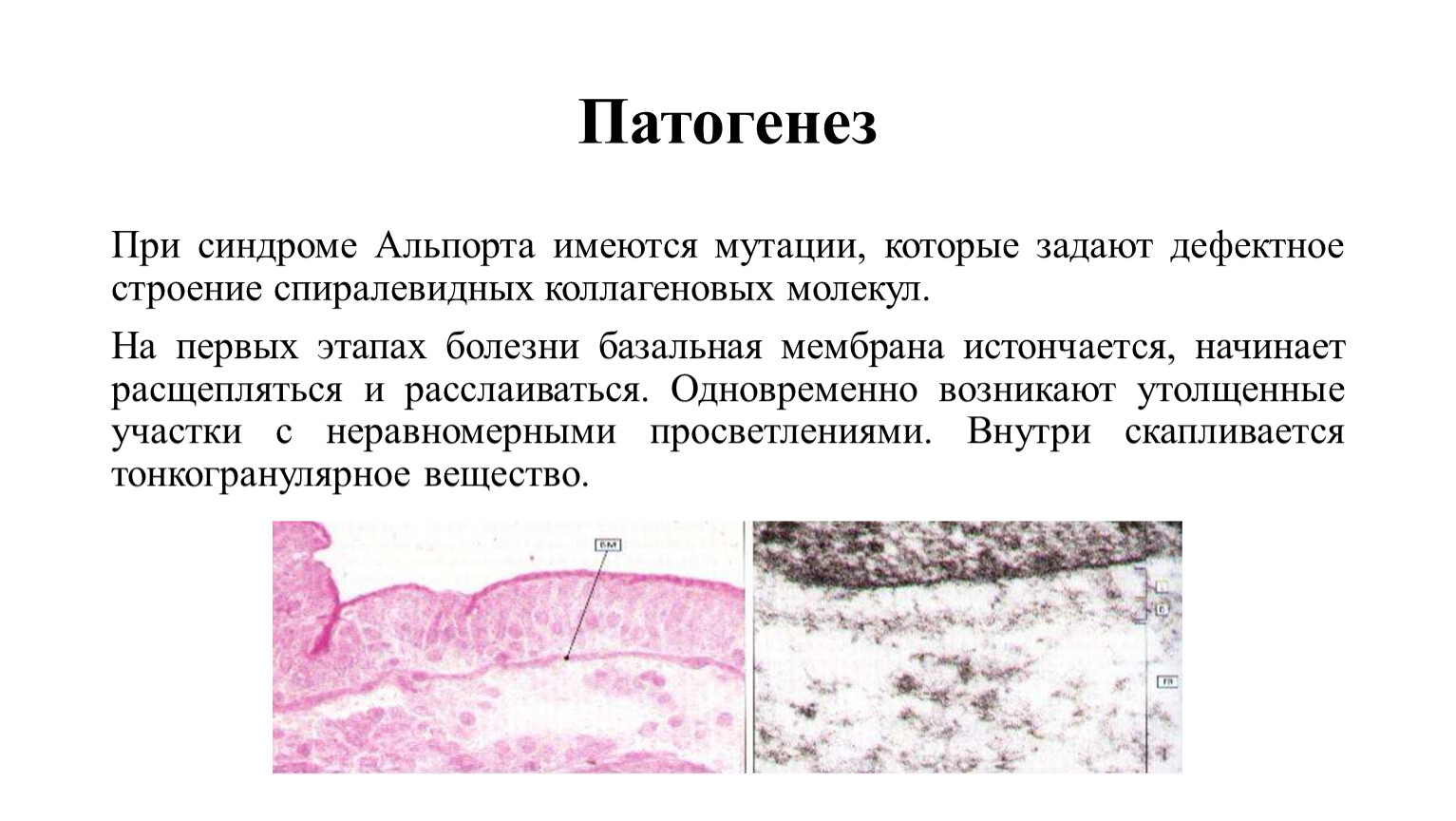
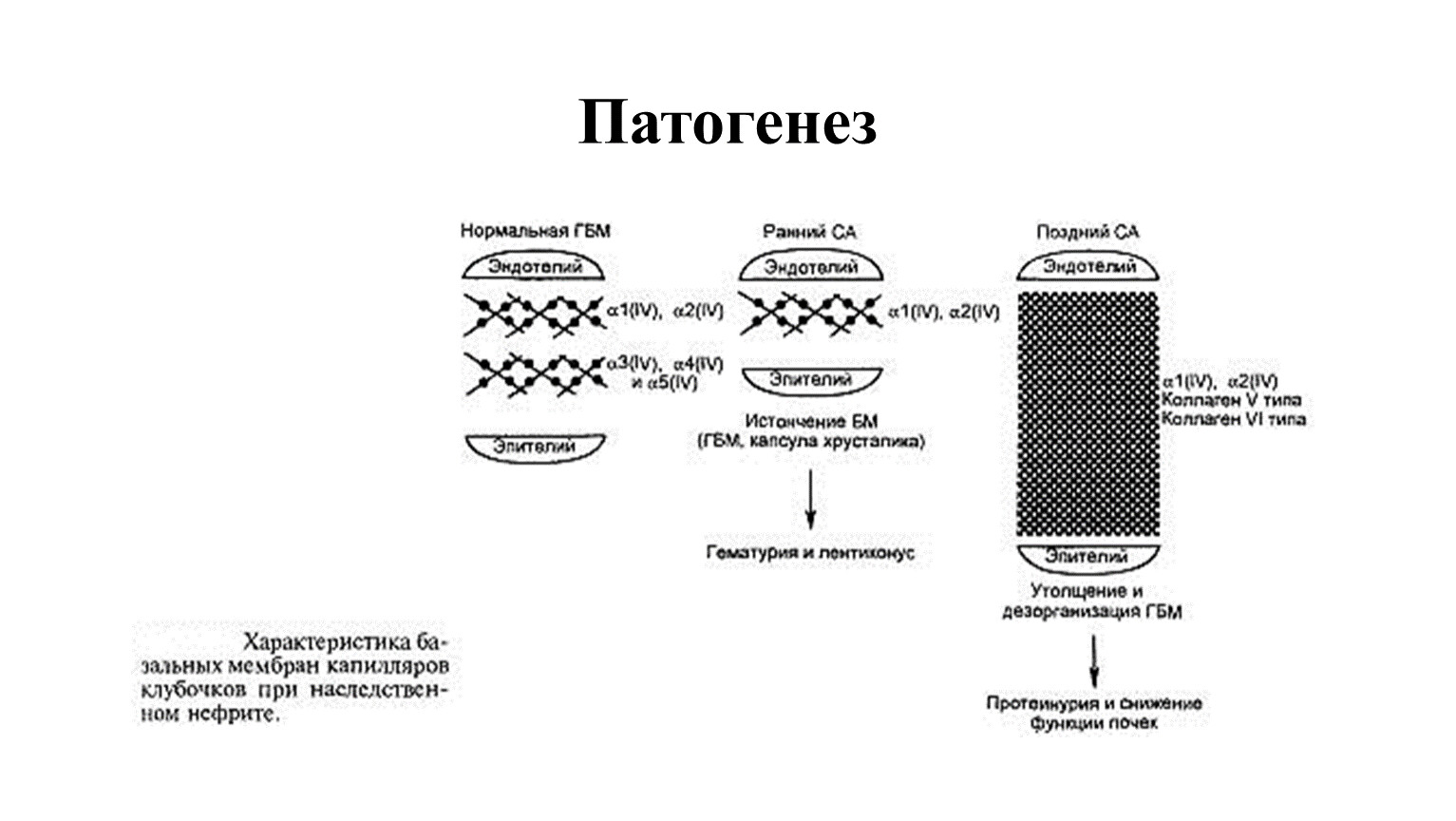
Патогенез
При синдроме Альпорта имеются мутации, которые задают дефектное строение спиралевидных коллагеновых молекул.
На первых этапах болезни базальная мембрана истончается, начинает расщепляться и расслаиваться. Одновременно возникают утолщенные участки с неравномерными просветлениями. Внутри скапливается тонкогранулярное вещество.

Патогенез
Прогрессирование болезни сопровождается полным разрушением базальной гломерулярной мембраны клубочковых капилляров, канальцев почек, структур внутреннего уха и глаз.
Таким образом, патогенетически синдром Альпорта представлен четырьмя звеньями:
мутацией гена,
дефектом строения коллагена,
деструкцией базальных мембран,
патологией почек (иногда – нарушением слуха и зрения).
Патогенез
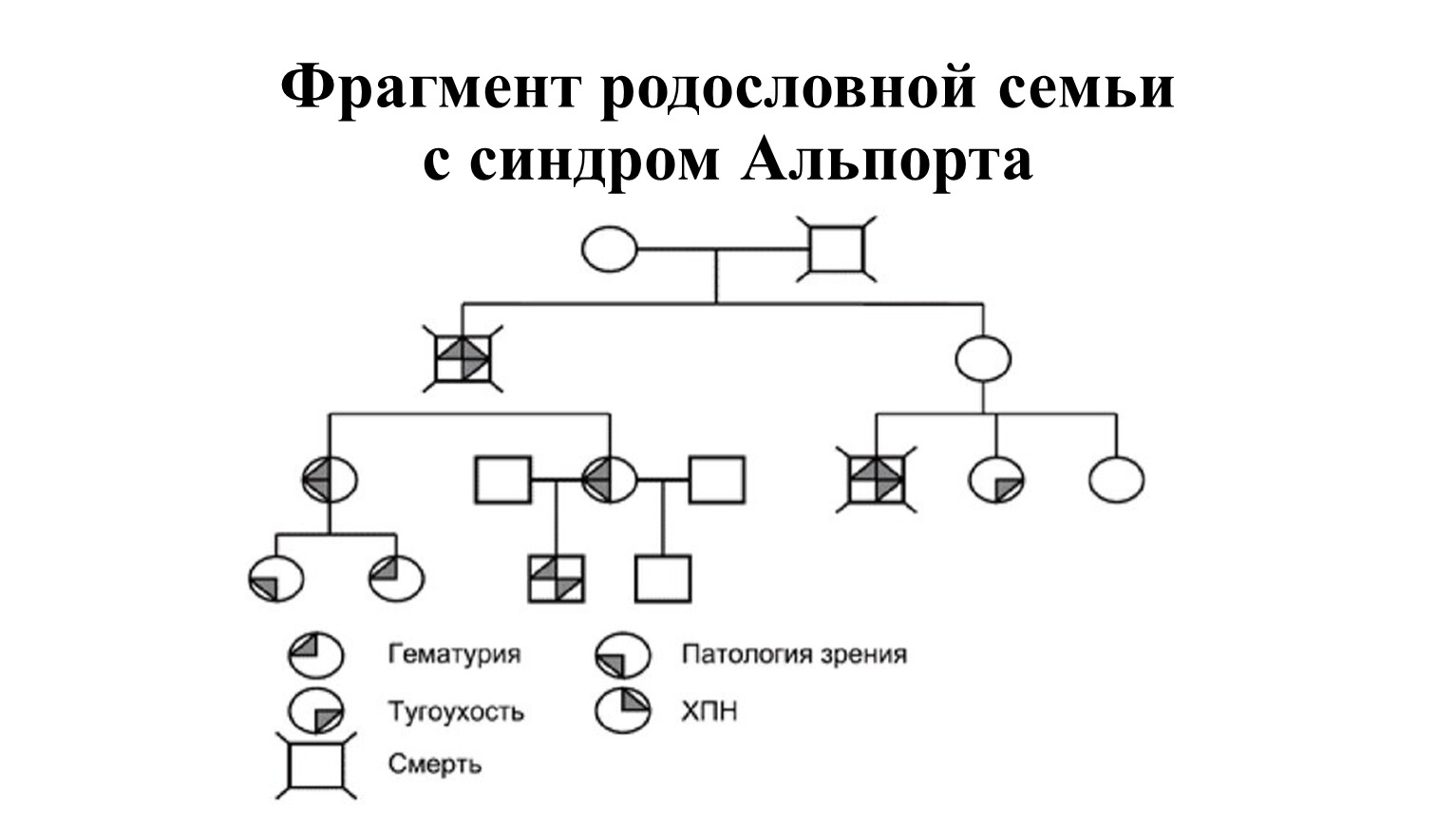
Фрагмент родословной семьи с синдром Альпорта
Симптомы
Самым распространенным проявлением синдрома Альпорта является гематурия. Микроскопически этот симптом определяется у 95% женщин и у 100% мужчин. При рутинном обследовании мальчиков гематурия обнаруживается уже в первые годы жизни.

Симптомы
Другой распространенный признак заболевания – протеинурия. Выведение белка с мочой у пациентов мужского пола с X-сцепленным синдромом начинается в раннем детском возрасте, у остальных – позже. У девочек и женщин уровень экскреции белка повышается незначительно, случаи выраженной протеинурии крайне редки. У всех больных отмечается неуклонное прогрессирование симптома.

Симптомы
Артериальная гипертензия характерна для мужчин с классическим типом синдрома и для пациентов обоих полов с аутосомно-рецессивным вариантом наследования.
У юношей, мужчин снижение функции почек достигает терминальной стадии к 16-35 годам, при медленном течении болезни – к 45-65 годам.
Иногда выявляются диффузные гладкомышечные опухоли пищевода и бронхов, проявляющиеся в позднем детстве дисфагией, рвотой, болями в эпигастрии и за грудиной, одышкой, частыми бронхитами.

Симптомы
Часто у больных формируется нейросенсорная тугоухость. Нарушения слуха дебютируют в детстве, но становятся заметными в подростничестве или молодости. У детей тугоухость распространяется только на звуки высокой частоты, обнаруживается в специально созданных условиях – при аудиометрии.
По мере взросления и прогрессирования синдрома нарушается слуховое восприятие средних и низких частот, в том числе человеческой речи.
При X-связанном синдроме расстройство слуха к 25 годам имеется у 50% больных мужчин, к 40 годам – у 90%.
Тяжесть тугоухости вариабельна, от изменений только в результатах аудиограммы до полной глухоты. Патологии вестибулярного аппарата отсутствуют.
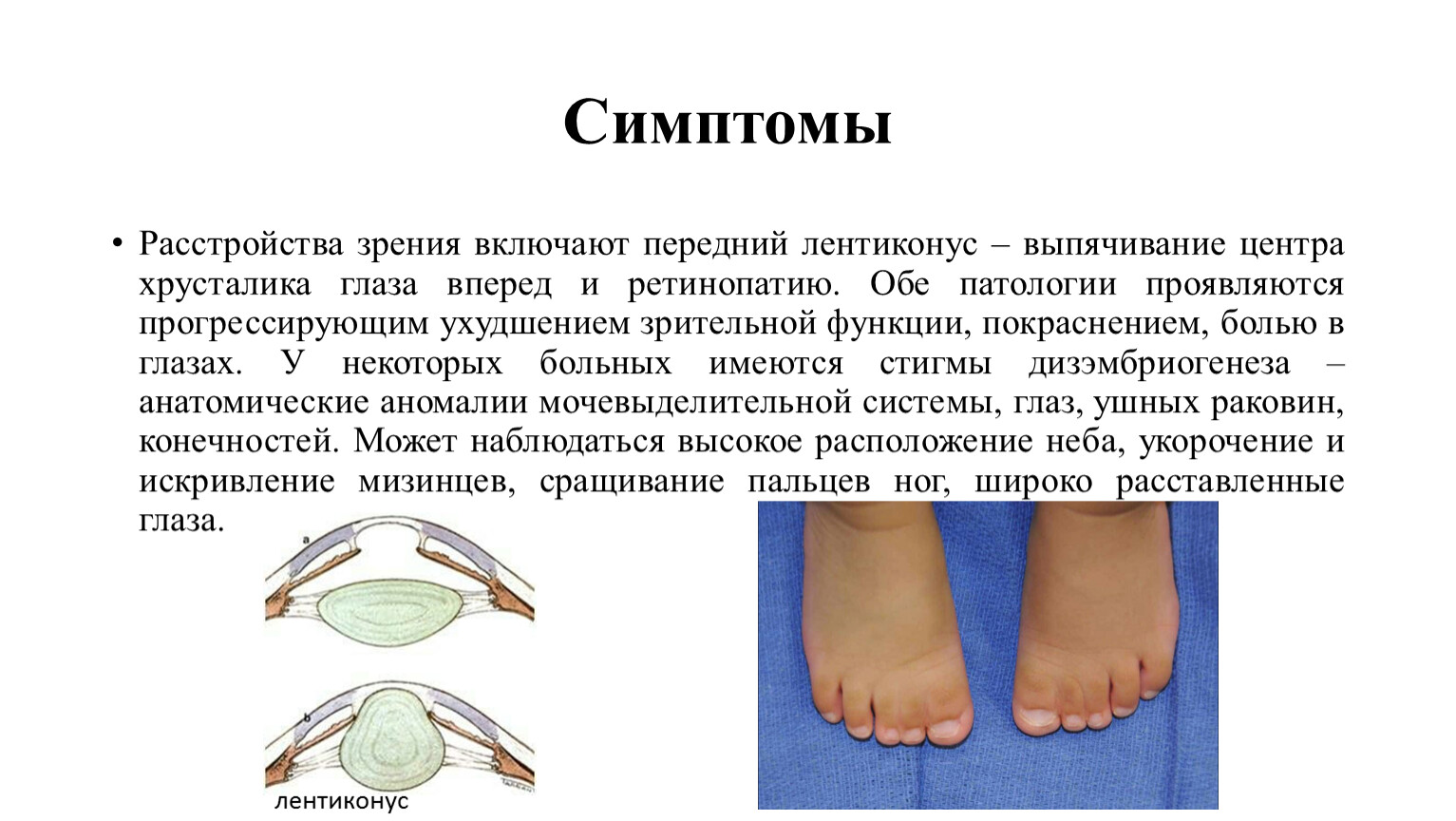
Симптомы
Расстройства зрения включают передний лентиконус – выпячивание центра хрусталика глаза вперед и ретинопатию. Обе патологии проявляются прогрессирующим ухудшением зрительной функции, покраснением, болью в глазах. У некоторых больных имеются стигмы дизэмбриогенеза – анатомические аномалии мочевыделительной системы, глаз, ушных раковин, конечностей. Может наблюдаться высокое расположение неба, укорочение и искривление мизинцев, сращивание пальцев ног, широко расставленные глаза.
лентиконус
Осложнения
Отсутствие лечения больных синдромом Альпорта приводит к быстрому прогрессированию глухоты и слепоты, формированию катаракт.
У части пациентов развивается полиневропатия – поражение нервов, сопровождающееся:
мышечной слабостью,
болями,
судорогами,
тремором,
парестезиями,
снижением чувствительности.
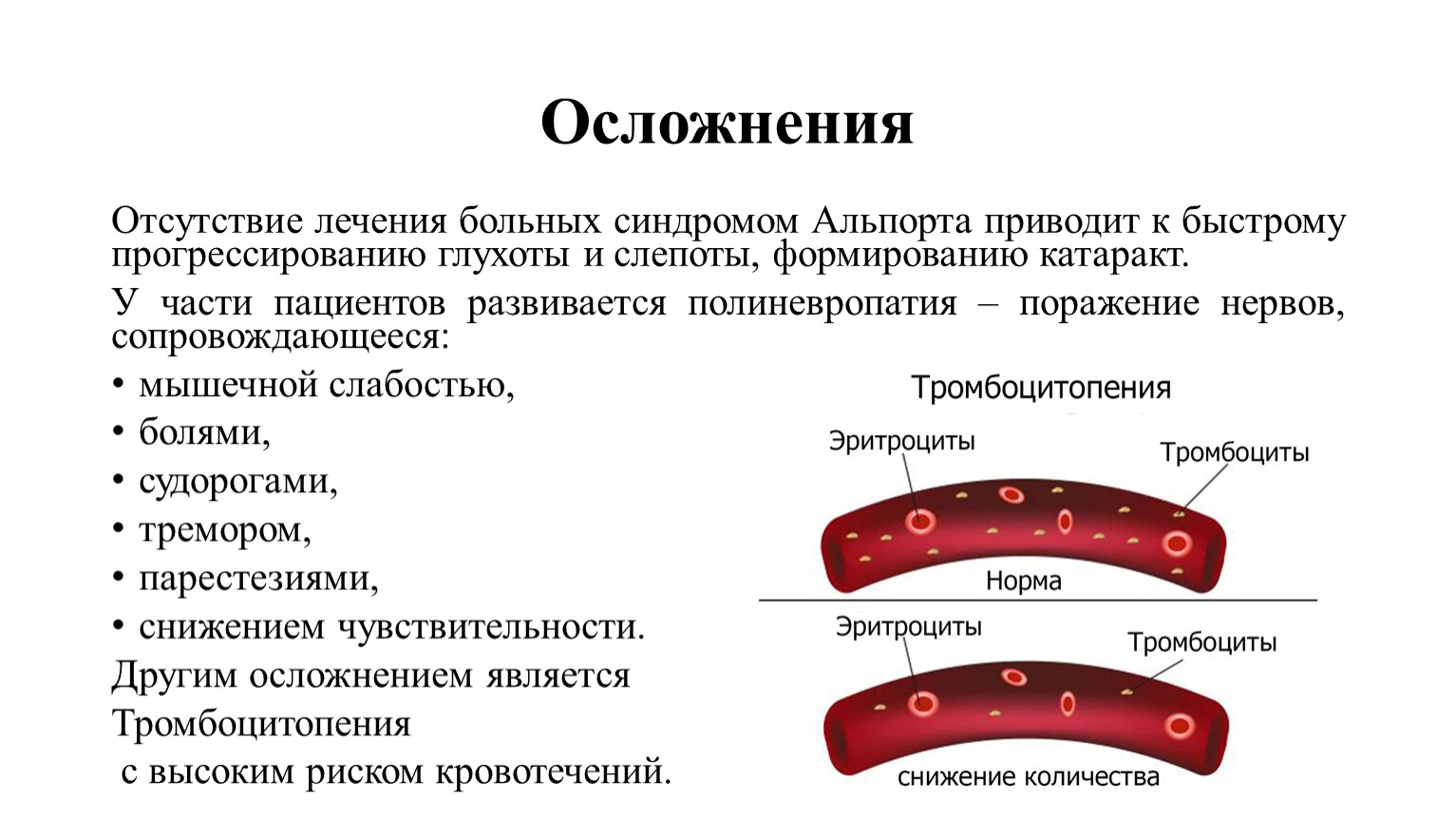
Другим осложнением является
Тромбоцитопения
с высоким риском кровотечений.

Осложнения
Наиболее опасным состоянием при наследственном нефрите считается терминальная стадия почечной недостаточности. Больше всего ей подвержены мужчины с типом наследования, сцепленным с половой X-хромосомой.
К 60 годам 100% больных этой группы нуждаются в процедурах:
гемодиализа (очищение крови),
перитонеального диализа,
трансплантации донорской почки.

Диагностика
В диагностическом процессе принимают участие врачи-нефрологи, урологи, терапевты и генетики.
При опросе выясняется возраст дебюта симптомов, наличие у родственников первой линии гематурии, протеинурии или смертельных исходов вследствие ХПН.
Для синдрома Альпорта характерно раннее начало и отягощенный семейный анамнез. Дифференциальная диагностика направлена на исключение гематурической формы гломерулонефритов, вторичных нефропатий.

Диагностика
Для подтверждения диагноза проводятся следующие процедуры:
Физикальное обследование. Определяется бледность кожных покров и слизистых оболочек, сниженный мышечный тонус, внешние и соматические признаки дизэмбриогенеза – высокое небо, аномалии строения конечностей, увеличенное расстояние между глазами, сосками. На ранних стадиях болезни диагностируется артериальная гипотония, на поздних – артериальная гипертония.
Общий анализ мочи. Обнаруживаются эритроциты и повышенное содержание белка – признаки гематурии и протеинурии. Показатель белка мочи напрямую коррелирует с тяжестью синдрома, по его изменению оценивается прогрессирование патологии, вероятность нефротического синдрома, ХПН. Возможно наличие признаков лейкоцитурии абактериального характера.
Диагностика
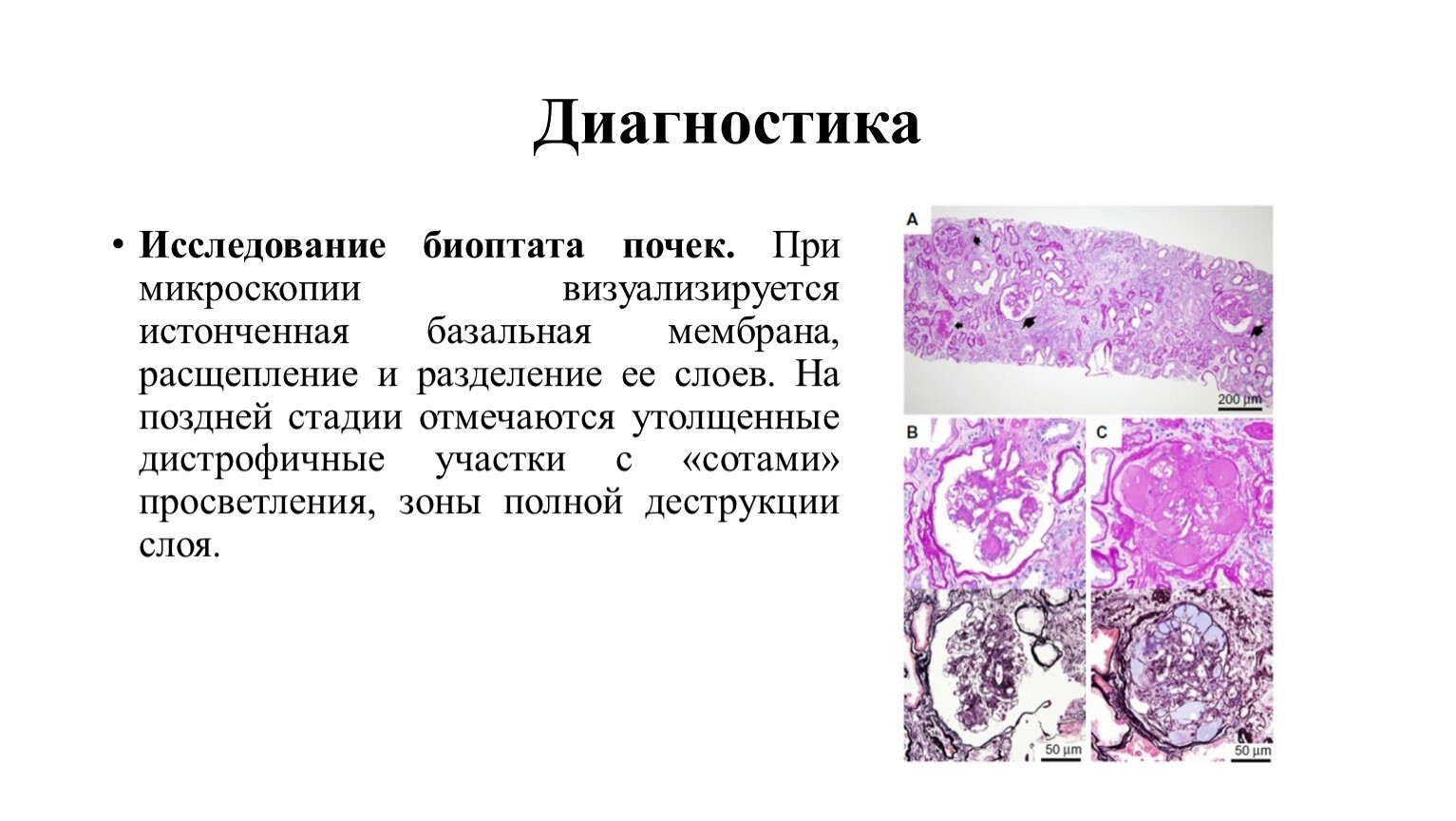
Исследование биоптата почек. При микроскопии визуализируется истонченная базальная мембрана, расщепление и разделение ее слоев. На поздней стадии отмечаются утолщенные дистрофичные участки с «сотами» просветления, зоны полной деструкции слоя.
Диагностика
Молекулярно-генетическое исследование. Генетическая диагностика не является обязательной, но позволяет составить более точный прогноз, подобрать оптимальную схему лечения. Изучается строение генов, мутации в которых обуславливают развитие синдрома. У большей части больных выявляются мутации гена COL4A5.

Диагностика
Аудиометрия, офтальмологическое исследование. Дополнительно пациентам могут быть назначены диагностические консультации сурдолога и офтальмолога.
При аудиометрии обнаруживается снижение слуха:
в детском и подростковом возрасте – билатеральная высокочастотная тугоухость,
во взрослом возрасте – низкочастотная и среднечастотная тугоухость.
Офтальмолог определяет искажение формы хрусталика, поражение сетчатки, наличие катаракты, снижение зрения.

Лечение синдрома Альпорта
Специфическая терапия отсутствует.
С раннего возраста проводится активное симптоматическое лечение, снижающее протеинурию. Оно позволяет предотвратить поражение и атрофию почечных канальцев, развитие интерстициального фиброза.
С помощью ингибиторов ангиотензинпревращающего фермента и блокаторов рецепторов к ангиотензину II удается приостановить прогрессирование заболевания, добиться регрессии гломерулосклероза, тубулоинтерстициальных и сосудистых изменений в почках. Пациентам с терминальной стадией ХПН (хроническая почечная недостаточность) назначается гемодиализ, перитонеальный диализ, решается вопрос о целесообразности трансплантации почек.

Методы терапии
Лечение синдрома Альпорта включает в себя диету, прием лекарственных средств, своевременную санацию очагов инфекции.
Прививки детям противопоказаны, вакцинация возможна только при наличии строгих показаний.
На данный момент не существует фармакологических препаратов, которые бы воздействовали на генетический дефект.
Широко применяют метаболические препараты, которые позволяют повысить.
При пониженном артериальном давлении у детей, следует принимать минимальные дозы лекарств, чтобы снизить скорость прогрессии недостаточности почек.
Материалы на данной страницы взяты из открытых источников либо размещены пользователем в соответствии с договором-офертой сайта. Вы можете сообщить о нарушении.