Поделиться

Тема: Хромосомные болезни
1. Хромосомные болезни.
Хромосомные болезни — это большая группа врожденных патологических состояний с множественными врожденными пороками развития, причиной которых является изменение количества или структуры хромосом. Возникают они в результате мутаций в половых клетках одного из родителей. Тогда все клетки организма больного имеют аномальный кариотип. Из поколения в поколение передаются нс более 3— 5% из них.
Однако изменение набора хромосом может произойти и во время первых делений зиготы, даже если она была сформирована из нормальных гамет. В таких случаях образуется мо- заицизм, т. е. часть клеток организма имеет другой набор хромосом.
Клиническое описание этих заболеваний появилось сше до открытия их хромосомной природы. Наиболее часто встречающаяся болезнь, трисомия 21, особый вид умственной отсталости у детей, была описана в 1866 г. английским педиатром Дауном и получила название «синдрома Дауна». Н.А. Ше- рсшсвским в 1925 г. дано первое клиническое описание синдрома моносомии по Х-хромосоме, а затем Г. Тернер в 1938 г. описал этот синдром. По фамилии этих ученых миосомию называют синдром Шсрсшсвского -Тернера. Г. Клайнфель- тср впервые описал аномалии в системе половых хромосом у мужчин — трисомия ХХУ. Изучение хромосомных болезней активно началось с 60-х гг. XX в. благодаря широкому развертыванию цитогенетических исследований, когда полностью сложилась клиническая цитогенетика.
Учение о хромосомной патологии сложилось в результате интенсивного изучения хромосом человека и хромосомных болезней.
Была показана роль хромосомных и геномных мутаций в патологии человека, расшифрована хромосомная причина многих синдромов врожденных пороков развития. В настоящее время описано около 1 000 различных видов аномалий хромосом у человека. Примерно 100 форм имеют клинически очерченную картину и называются синдромами.
Распространенность хромосомных болезней одинакова во всех национальных и этнических группах и составляет в среднем 7—8 больных на каждую 1 000 новорожденных. В России эта патология регистрируется примерно у 12 000 новорожденных ежегодно.
Хромосомные аберрации у плода часто являются причиной неблагополучного исхода беременности. Около 90% эмбрионов человека, имеющих эти аномалии, погибают еще внутриутробно. Примерно 50% всех диагностированных самопроизвольных абортов обусловлены хромосомными нарушениями. Подобные нарушения выявляются у 7% мертворожденных. Примерно 45% всех случаев множественных врожденных пороков у детей составляют хромосомные синдромы.
Различают геномные синдромы и структурные изменения хромосом.
Геномные синдромы характеризуются изменением числа хромосом. У человека обнаружены только три типа геномных мутаций: тетраплоидия, триплоидия и анеуплоидия.
Полиплоидия редко обнаруживается у новорожденных, у которых зарегистрированы случаи триплоидии (69 хромосом) и тетраплоидии (92 хромосомы). Это нарушение хромосомного набора чаще выявляется у эмбрионов при выкидышах в первом триместре беременности.
Анеуплоидия — это увеличение или уменьшение числа хромосом, не кратное гаплоидному. Чаще всего у человека регистрируется наличие дополнительной хромосомы — трисомия по аутосомам. При этом какая-либо хромосома представлена в организме тремя копиями, а кариотип включает 47 хромосом. Возможно и больше копий (4 или 5) одной хромосомы в организме. Увеличиваться может число как аутосом, так и половых хромосом. Отсутствие одной хромосомы называется моносомией. Встречаются полисомии по половым хромосомам — три-, тетра-, пентасомии. Кариотип человека в этом случае содержит 45 хромосом. Совместимой с жизнью является только моносомия по Х-хромосоме (синдром Шерешевс- кого —Тернера).
Все хромосомные болезни принято делить на две группы:
1) Связанные с аномалиями числа хромосом. В эту группу входят три подгруппы:
— болезни, причиной которых является нарушение числа хромосом;
— болезни, связанные с увеличением или уменьшением числа половых X- и У-хромосом;
— болезни, обусловленные полиплоидией — кратным увеличением гаплоидного набора хромосом.
2) Связанные со структурными нарушениями (аберрациями) хромосом.
Их причинами являются:
— Транслокации — обменные перестройки между нсгомо- логичными хромосомами.
— Делеции — потери участка хромосомы.
— Инверсии — повороты участка хромосомы на 180°.
— Дупликации — удвоения участка хромосомы.
— Изохромосомия — хромосомы с повторяющимся генетическим материалом в обоих плечах.
— Возникновение кольцевых хромосом — соединение двух концевых делеций в обоих плечах хромосомы
2. Синдромы с числовыми аномалиями аутосом (синдром Дауна, синдром Эдвардса, синдром Патау).
Синдром Дауна (трисомия 21) — часто встречающееся и хорошо изученное хромосомное заболевание. Впервые оно было описано в 1866 г. английским педиатром А. Дауном. В 1959 г. J. 1^е]еппе с соавторами установили, что причиной синдрома Дауна является наличие дополнительной 21 -й хромосомы в кариотипе больного (рис. 8.4).
Встречается у новорожденных с частотой — 1 больной на 700-800 новорожденных. Частота рождения детей с болезнью Дауна зависит от возраста женшин-матерей (до 18 лет и старше 35 лет). 80—90% всех случаев заболевания являются результатом нсрасхождсния хромосом в мейозе у матери или дробления зиготы. Но остальные 10-20% трисомий 21-й хромосомы вызваны нарушениями сперматогенеза у отца больного.
Синдром Дауна может возникнуть в результате разных вариантов изменений хромосом, приводящих к увеличению числа 21-й хромосомы:
1) Основную долю (94%) составляют случаи простой полной трисомии 21-й хромосомы. Кариотип таких больных можно записать следующим образом: 47, ХУ, +21 или 47, XX, +21.
2) Около 2% больных детей имеют мозаичные варианты синдрома Дауна. При этом часть клеток организма больного содержит дополнительную хромосому, а другие клетки имеют нормальный кариотип. Мозаичные варианты синдрома Дауна обычно сопровождаются менее выраженными изменениями фенотипа по сравнению с простой трисомией.
3) Почти 50% транслокационных форм наследуются от ро- дителей-носителей, а 50% транслокаций возникают вновь. Иногда заболевание возникает при транслокации небольшого участка длинного плеча 21-й хромосомы. Эта форма заболевания регистрируется примерно в 4% случаев.
Клинические проявления синдрома Дауна разнообразны и заметны уже при рождении (рис. 8.5).
Для синдрома Дауна характерны: форма головы с уплощенным затылком; толстая кожная складка на задней поверхности шеи; лоб скошенный и узкий; лицо плоское; переносица широкая и вдавленная; язык у больных большой и виден между губами; постоянно открытый рот; толстые губы; «монголоидный» разрез глаз; типичен эпикант; ушные раковины уменьшены и деформированы. Со стороны костно-мышечной
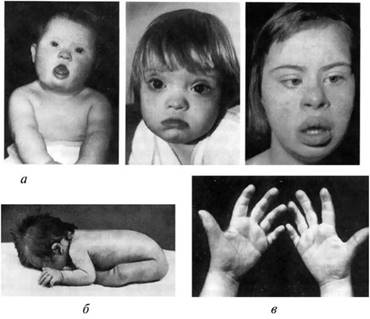
Рис. 8.5: а — дети разного возраста с характерными чертами синдрома Дауна (брахицефалия, круглое лицо, макроглоссия и открытый рот, эпикант, гипертелоризм, широкая переносица, «карпий рот», косоглазие); б — резкая гипотония у пациента с синдромом Дауна; в — ладони взрослого мужчины с синдромом Дауна (усиленная морщинистость, на левой руке четырехпальцевая, или «обезьянья», складка) системы характерны: низкий рост; короткая шея; воронкообразная или килевидная грудина; широкие кисти и стопы с короткими пальцами; глубокий борозды на ладонях; первый палеи на стопах широко отстоит от других пальцев — «сандалевидный промежуток»; мышечная гипотония с разболтанностью суставов.
При синдроме Дауна около 50% больных имеют врожденные пороки сердца. Обычно это дефекты межпредсердной или межжелудочковой перегородок.
Для всех больных этим синдромом характерна умственная отсталость — дебильность в 75% случаев, имбецильность у 20% больных, идиотия — в 5% случаев. При синдроме Дауна отмечается задержка физического и умственного развития, формирования моторных навыков и речи. Дети позже начинают ходить и говорить. У них резко нарушено абстрактное мышление. Они легче осваивают навыки, связанные с физическими движениями, чем речевые. Дети с синдромом Дауна внимательные, ласковые, послушные и общительные, терпеливые при обучении.
Продолжительность жизни при синдроме Дауна короче, чем у здоровых людей. Врожденные пороки внутренних органов, сниженная приспособляемость детей с синдромом Дауна часто приводят к смерти в первые 5 лет. Такие дети значительно чаще страдают острыми инфекциями и злокачественными заболеваниями крови. Средняя продолжительность жизни больных составляет 20 лет.
Лечение больных с синдромом Дауна должно быть комплексным и нсспепифичным:
1) Развитие моторных навыков и всех органов чувств: зрения, слуха, осязания, обоняния.
2) Полноценное питание, развивающие занятия, общеукрепляющие мероприятия — массаж и гимнастика.
3) Стимуляция двигательной активности ребенка — в течение дня несколько раз поворачивают на животик, при этом под грудь подкладывают небольшую подушечку. В возрасте от 2 до 6 месяцев необходимо поворачивать ребенка на бочок и животик.
4) Использование ноотропных лекарственных средств, укрепляющих ЦНС.
5) В возрасте от 6 до 12 месяцев необходимо обучать ребенка присаживаться и самостоятельно сидеть.
Многие больные с трисомией 21 способны жить самостоятельно, создавать семьи, овладевать несложными профессиями. С помощью специальных методов обучения, укрепления здоровья, правильного питания и ухода, проведения необходимого лечения можно продлить жизнь таким больным.
Синдром Патау (трисомия по 13-й хромосоме) — был впервые описан в 1960 г. К. Патау с соавторами у детей с множественными пороками развития (рис. 8.6). Синдром Патау оказался вторым патологическим состоянием человека, при котором были установлены изменения хромосом. Это заболевание встречается у детей с частотой в среднем 1 больной на 6 000 новорожденных. Мальчики и девочки поражаются одинаково часто. Часть детей с синдромом Патау погибают в пренатальном периоде.
80—85% всех случаев заболевания обусловлены нерасхож- денисм хромосом в мейозе в процессе формирования половых клеток родителей, т. е. это является результатом спонтанной
. Вероятность возникновения таких мутаций увеличивается с возрастом матери, как и при синдроме Дауна. Кариотип таких больных: 47, XX, +13 или 47, ХУ, +13.
Примерно 15% случаев синдрома Патау являются результатом транслокации 13-й хромосомы на какую-нибудь из хромосом группы Э. Очень редко встречаются другие цитологические случаи этого синдрома: мозаицизм, изохромосомы, другие транслокации и т. д.
Хромосома 13 значительно крупнее 21-й хромосомы, и, соответственно, ес трисомия вызывает значительно более тяжелые структурные и функциональные нарушения в организме ребенка.
Для беременности таким плодом характерны многоводие и угроза выкидыша. Масса тела новорожденного с синдромом Патау ниже нормального веса.
Синдром Патау сопровождается множественными врожденными пороками развития головного мозга и лица. Окружность черепа уменьшена, что приводит к формированию микроцефалии. В теменной области волосистой части головы часто выявляется участок отсутствия кожи до 1 см в диаметре. Лоб скошенный, глазные щели узкие, переносица запавшая, глаза недоразвиты (микрофтальмия) с помутнением роговицы, ушные раковины расположены низко и деформированы. Типичным признаком синдрома Патау у всех больных являются расщелины верхней губы и нёба, часто двухсторонние (рис. 8.7).
Всегда обнаруживаются пороки нескольких наружных и внутренних органов: полидактилия на руках, и чаше двухсторонняя, дефекты перегородок сердца, аномалии мочеполовых органов, дефекты поджелудочной железы и печени. Типичными для детей с трисомией 13-й хромосомы являются пороки развития половых органов: неопущение яичек (криптор- хизм) и недоразвитие полового члена у мальчиков, удвоение матки и влагалища у девочек. При подозрении на синдром Патау показано УЗИ всех внутренних органов.
В связи с тяжелыми врожденными пороками развития большинство детей с синдромом Патау живут недолго и 95% таких больных умирают до 1-го года жизни. Однако некоторые
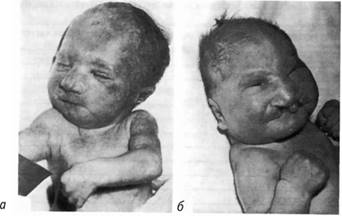
Рис. 8.7. Новорожденные с синдромом Патау: б — тригоноцефалия; двухсторонняя расщелина верхней губы и нёба; узкие глазные щели; низко расположенные; а — деформированные ушные раковины; микрогения; флексорное положение кистей
больные с синдромом Патау живут несколько лет. У таких больных выражена задержка психомоторного развития, имеется идиотия.
Лечение детей с синдромом Патау неспецифическое. Проводятся операции по поводу врожденных пороков развития, общеукрепляющее лечение, профилактика инфекционных и простудных заболеваний. Тщательный уход за такими пациентами облегчает их состояние, предупреждает инфекционные осложнения. Количество и состав основных ингредиентов пищи для таких больных должны соответствовать их возрасту.
Синдром Эдвардса (трисомия 18) был описан в 1960 г. Дж. Эдвардсом. Почти во всех случаях синдром Эдвардса обусловлен регулярной трисомией 18-й хромосомы. Кариотип больных при этом заболевании: 47, XX, +18 или 47, ХУ, + 18 (рис. 8.8).
Мозаицизм и транслокационные формы встречаются редко. Частота больных среди новорожденных — 1 больной ребенок на 7 000 новорожденных. Соотношение девочек и мальчиков с синдромом Эдвардса составляет 1 : 3.
Беременность при синдроме Эдвардса также осложняется угрозой прерывания и многоводием. Весьма характерно несоответствие размеров плода сроку беременности. При рождении дети имеют очень низкую массу тела, в среднем 2 170 г при доношенной беременности. При синдроме Эдвардса отмечается выраженная задержка пренатального развития при нормальной продолжительности беременности.
Синдром Эдвардса — это множественные врожденные пороки развития лицевой части головы, сердца, костной системы, половых органов. Чаше всего регистрируются дефекты развития конечностей, недоразвитие больших пальцев рук и лучевых костей, неправильно сформированные стопы с выступающей пяткой и провисанием свода («стопа-качалка»), укорочение первой плюсневой кости. Иногда обнаруживаются спинномозговые грыжи и расщелины верхней губы, недоразвитие глаз — микрофтальмия.
Череп имеет долихоцефалический, выступающий затылок, глазные щели короткие, нижняя челюсть и отверстие рта маленькие, челюсть скошена назад, ушные раковины деформированы и расположены низко, слуховой проход узкий, грудина укорочена, грудная клетка широкая. Отмечается флексорнос положение кистей рук. Имеется аномальная стопа — пятка выступает, а свод стопы провисает, первый палец стоп короче второго пальца. Мышечный тонус у таких больных обычно повышен. Дети лежат в кроватке, отведя голову назад, с согнутыми конечностями. У большинства пациентов с синдромом Эдвардса определяются пороки сердца и чаще всего — дефект межжелудочковой перегородки.
Часто выявляются пороки развития желудочно-кишечного тракта: атрезия (отсутствие отверстия) пищевода, незавершенный поворот кишечника и т. д. У таких детей отмечаются недоразвитие (гипоплазия) легких, сращение почек, удвоение мочеточников, неопушение яичек (крипторхизм) у мальчиков (рис. 8.9).
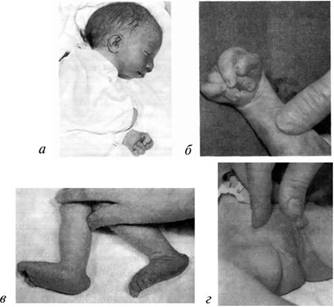
Рис. 8.9: а — новорожденный с синдромом Эдвардса (выступающий затылок, микрогения, флексорное положение кисти); б — характерное для синдрома Эдвардса положение пальцев (возраст ребенка 2 месяца); в — стопа-качалка (пятка выступает, свод провисает); г — гипогенитализм у мальчика (крипторхизм, гипоспадия)
Дети с синдромом Эдвардса умирают до 1 года от осложнений, связанных с врожденными пороками развития, таких как сердечно-сосудистая недостаточность, пневмония, кишечная непроходимость. Для больных старшего возраста характерна глубокая умственная отсталость. Уход за больными в основном заключается в предупреждении инфекционных осложнений.
3. Синдромы с числовыми аномалиями половых хромосом (синдром Шерешевского-Тернера, синдром Клайнфельтера, синдром трисомии Х).
Полисомии по половым хромосомам представляют собой большую группу хромосомных болезней, с различными комбинациями дополнительных X- или У-хромосом и комбинациями разных клонов в случае мозаицизма. Общая частота полисомии по X- или У-хромосомам среди новорожденных составляет 1,5 : 1000-2 : 1000. В основном это полисомии XXX, ХХУ и ХУУ. 25% составляют мозаичные типы полисомий по половым хромосомам.
В отличие от аутосомных трисомий клиническая картина этих патологических состояний характеризуется нарушением полового развития.
Синдромы полисомии Х-хромосомы включают в себя трисо- мию X (кариотип пациентки — 47, XXX), тетрасомию X (48, ХХХХ), пснтасомию X (49, ХХХХХ). Дополнительные Х-хромосомы у таких людей инактивируются, поэтому такие патологические состояния являются совместимыми с жизнью, несмотря на выраженный хромосомный дисбаланс.
Трисомия Х-хромосомы среди новорожденных девочек составляет 1 : 1000. Обычно такая патология обнаруживается при случайном массовом обследовании детей. Женщины с кариотипом XXX в полном или мозаичном варианте имеют нормальное физическое и психическое развитие. Объясняется это тем, что в клетках две Х-хромосомы гетерохроматинизирова- ны, а функционирует лишь одна, как у нормальной женщины. Однако при трисомии X с возрастом увеличивается риск возникновения психических заболеваний. Интеллектуальное развитие нормальное или на нижней границе нормы. У некоторых женщин имеются нарушения репродуктивной функции — аменорея, дисменорея, ранняя менопауза. Редко обнаруживаются другие патологические изменения: микроцефалия, косоглазие, сколиоз, высокий рост, нарушение менструальной функции и бесплодие.
Увеличение числа Х-хромосом в кариотипе сопровождается усугублением поражения нервной системы, формированием пороков развития и нарушением функции половых органов.
При тетрасомии и пентасомии X-хромосомы у пациенток отмечаются значительная умственная отсталость, судороги, недоразвитие гениталий, пороки развития конечностей (их маленькие размеры, сращение лучевой и локтевой костей), врожденные пороки сердца, необычный внешний вид. Варианты синдрома Х-полисомии без У-хромосомы с числом более 3 встречаются редко. С увеличением числа дополнительных Х-хромосом нарастают отклонения от нормы.
Диагностика полисомии Х-хромосомы основывается на исследовании кариотипа. Заключение об избыточном количестве Х-хромосом можно предварительно сделать на основании исследования полового хроматина. Снижение интеллекта от пограничной умственной отсталости до различных степеней олигофрении, черепно-лицевые дисформии, аномалии зубов, скелета и половых органов описаны у2/, больных женщин с тетра- и пснтасомией X.
Методика терапии при синдроме полисомии по Х-хромо- соме диктуется теми патологическими нарушениями, которые отмечаются у пациенток.
Синдром Шерешевского—Тернера (45, X) — это единственная форма у рожденных детей, с кариотипом 45, X, 90% зачатий абортируется самопроизвольно. Впервые этот синдром был описан русским ученым Н.А. Шсрешевским в 1925 г. В 1938 г. Г. Тернер дал полное описание этого заболевания, которое получило название синдрома Шерешевского— Тернера. Позже была установлена этиология заболевания — моносомия по Х-хромосоме у женщин.
Частота этого синдрома составляет 1 : 3 000 новорожденных девочек.
Чаще всего при этом заболевании регистрируется полная моносомия по Х-хромосоме (60% всех больных) (рис. 8.10), из них в 80—85% случаев имеет материнское происхождение, а в 15-20% — отцовское.
Кариотип больных при этом заболевании включает 45 хромосом с одной Х-хромосомой (45, X). Кроме истинной мо- носомии во всех клетках встречаются другие формы хромосомных аномалий по половым хромосомам. Среди них делеции короткого или длинного плеча Х-хромосомы 46, X, Хр-; 46, X, Хч~). Часто выявляются формы мозаицизма, примерно в 20% всех случаев.
Беременность плодом с этим заболеванием сопровождается выкидышем.
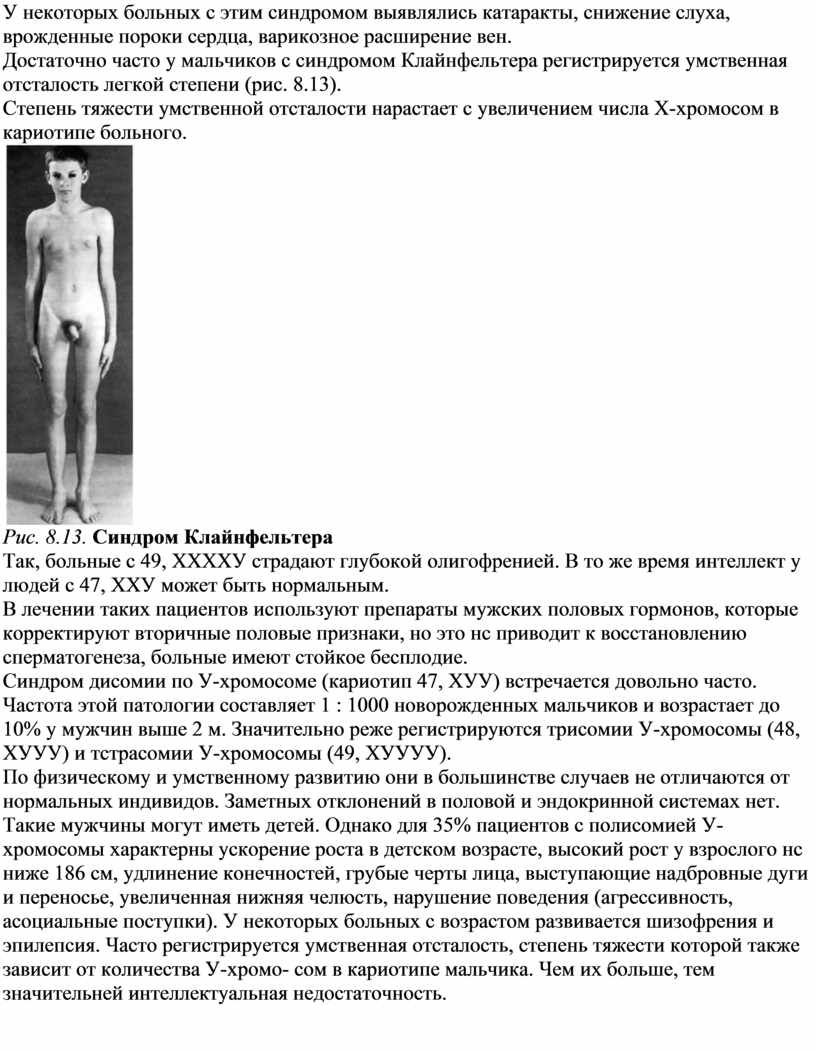
Для синдрома Шсрешсвско го—Тернера характерны: гипогонадизм, недоразвитие половых органов и вторичных половых признаков; врожденные пороки развития; низкий рост. Проявлением заболевания являются короткая шея с избытком кожи и крыловидными складками, лимфатический отек на тыльных поверхностях кистей рук, предплечий, стоп и голеней. Подобные изменения развиваются не у всех девочек с синдромом Шерешевского-Тернера. Часто в первые годы жизни у детей встречаются разные врожденные пороки сердца и почек, до 20% случаев. В подростковом возрасте можно заметить задержку роста ребенка. Необходимо учитывать, что 80% всех случаев низкорослое™ у девочек обусловлены синдромом Шерсшсвского—Тернера. Рост взрослых девушек с этим заболеванием составляет в среднем 135—140 см. Характерным для синдрома Шерсшсвского—Тернера является задержка полового развития у девочек 12—14 лет. Телосложение у таких пациентов коренастое. На коже часто расположено множество родинок. Подбородок несколько выступает вниз, уши расположены низко. Грудная клетка широкая, «шитообразная». Соски расставлены широко друг от друга — гипертелоризм сосков. Часто выявляются различные деформации скелета: воронкообразная грудная клетка, вальгусное положение предплечий и голеней и др. (рис. 8.11). Интеллектуальное развитие пациенток с этим заболеванием близко к норме. Однако для таких больных характерны инфантильность и снижение познавательных способностей.
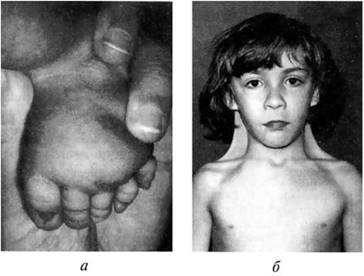
Рис. 8.11: а — лимфатический отек стопы у новорожденного с синдромом Шерешевского-Тернера, маленькие выпуклые ногти;
6 — девочка с синдромом Шерешевского-Тернера (шейные крыловидные складки, широко расположенные и недоразвитые соски молочных желез)
У взрослых с синдромом Шсрсшсвского—Тернера отмечают нарушение скелета, черепно-лицевые дисформии, валь- гусную девиацию коленных и локтевых суставов, остеопороз, бочкообразную грудную клетку, птоз, эпикант, рстрогению, бесплодие. Но было известно несколько случаев беременностей у женщин с полной моносомией по Х-хромосоме.
Лечение при этом заболевании комплексное. До 12—14 лет таким девочкам проводится терапия, корригирующая задержку роста. С 13—14 лет проводится гормональное лечение препаратами женских половых гормонов, которые вызывают формирование вторичных половых признаков и менструального цикла. Оперативно исправляются врожденные пороки развития. Современные методы гормональной терапии и экстракорпорального оплодотворения с использованием донорской яйцеклетки уже дали возможность рождения здорового ребенка нескольким женщинам, имеющим моносомию по Х-хро- мосоме. Проводится психотерапия. Своевременное применение всех методов лечения, применение генно-инженерного гормона роста обеспечивают почти полную компенсацию патологических проявлений у этих больных.
Синдром Клайнфельтера — наиболее часто встречающийся с типичной картиной синдром с набором 47, ХХУ. Болеют только мальчики. Включает случаи полисомии по половым хромосомам в кариотипе у мужчины, при которых имеется не менее двух Х-хромосом и не менее одной У-хромосомы. Впервые был описан в 1942 г. (рис. 8.12). Частота синдрома Клайн- фельтсра в полном и мозаичном вариантах — 1 больной ребенок на 500-700 новорожденных. Около 80% всех случаев заболевания связаны с присутствием в кариотипе дополнительной Х-хромосомы (47, ХХУ). У других пациентов с синдромом Клайнфельтера в основном регистрируются мозаичные формы, когда, например, часть клеток организма больного может содержать нормальный набор хромосом.
Характерная клиническая картина формируется примерно к 12—15-летнему возрасту, когда обычно впервые обнаруживается недоразвитие яичек и вторичных мужских половых признаков, увеличиваются молочные железы — гинекомастия.
Вторичные половые признаки выражены слабо. Телосложение — узкие плечи, широкий таз. В резко уменьшенных яичках нс происходит сперматогенез (азооспермия, олигоспермия). Нарушение развития гениталий сопровождается стойким бесплодием.
Больные с синдромом Клайнфельтера высокого роста. Часто встречаются диспропорционально длинные нижние конечности, сколиоз, деформация грудной клетки. У некоторых больных с этим синдромом выявлялись катаракты, снижение слуха, врожденные пороки сердца, варикозное расширение вен.
Достаточно часто у мальчиков с синдромом Клайнфельтера регистрируется умственная отсталость легкой степени (рис. 8.13).
Степень тяжести умственной отсталости нарастает с увеличением числа Х-хромосом в кариотипе больного.
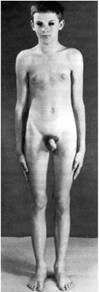
Рис. 8.13. Синдром Клайнфельтера
Так, больные с 49, ХХХХУ страдают глубокой олигофренией. В то же время интеллект у людей с 47, ХХУ может быть нормальным.
В лечении таких пациентов используют препараты мужских половых гормонов, которые корректируют вторичные половые признаки, но это нс приводит к восстановлению сперматогенеза, больные имеют стойкое бесплодие.
Синдром дисомии по У-хромосоме (кариотип 47, ХУУ) встречается довольно часто. Частота этой патологии составляет 1 : 1000 новорожденных мальчиков и возрастает до 10% у мужчин выше 2 м. Значительно реже регистрируются трисомии У-хромосомы (48, ХУУУ) и тстрасомии У-хромосомы (49, ХУУУУ).
По физическому и умственному развитию они в большинстве случаев не отличаются от нормальных индивидов. Заметных отклонений в половой и эндокринной системах нет. Такие мужчины могут иметь детей. Однако для 35% пациентов с полисомией У-хромосомы характерны ускорение роста в детском возрасте, высокий рост у взрослого нс ниже 186 см, удлинение конечностей, грубые черты лица, выступающие надбровные дуги и переносье, увеличенная нижняя челюсть, нарушение поведения (агрессивность, асоциальные поступки). У некоторых больных с возрастом развивается шизофрения и эпилепсия. Часто регистрируется умственная отсталость, степень тяжести которой также зависит от количества У-хромо- сом в кариотипе мальчика. Чем их больше, тем значительней интеллектуальная недостаточность.







Материалы на данной страницы взяты из открытых источников либо размещены пользователем в соответствии с договором-офертой сайта. Вы можете сообщить о нарушении.
