Поделиться

ЛЕКЦИЯ
СЕРДЕЧНАЯ НЕДОСТАТОЧНОСТЬ
Недостаточность кровообращения
Под недостаточностью кровообращения понимают состояние, при котором сердечно-сосудистая система не обеспечивает потребности тканей и органов в кровоснабжении, т.е. доставку к ним с кровью кислорода, питательных веществ и удаления из них углекислоты и продуктов обмена.
(НК) - неспособность сердечно-сосудистой системы снабжать органы и ткани необходимым для их нормального функционирования количеством крови, как в покое, так и при предъявлении к организму повышенных требований (физическая или эмоциональная нагрузка, интеркуррентные заболевания), то есть состояние системы кровообращения, при котором развивается генерализованная циркуляторная гипоксия.
НК развивается в результате:
- нарушения нейрогуморальной регуляции сосудистого тонуса;
- поражения сосудистой стенки;
- гиповолемии;
- нарушения реологических свойств крови.
НК может быть компенсированной, если проявляется только при нагрузке и некомпенсированной, если проявляется в покое.
Выделяют сердечную недостаточность и сосудистую недостаточность.
НК может быть острой и хронической.
К острой недостаточности кровообращения относят острую сердечную лево и правожелудочковую недостаточность, и острую сосудистую недостаточность (шок, обморок, коллапс).
Хроническая недостаточность развивается медленно и может быть связана как с заболеваниями сердца.
Функцией сердца является ритмическое нагнетание в артерии крови, притекающей к нему из вен. Эта функция выполняется благодаря попеременным ритмическим сокращениям и расслаблением мышечных волокон, образующих стенку предсердий и желудочков.
Сокращение сердца координируется специализированными клетками, образующими синусовый и предсердно-желудочковый узлы, а также внутрисердечным пучком Гиса и волокнами Пуркинье.
Ударный объем – это количество крови, изгоняемой из желудочков сердца во время систолы. Сердечный выброс крови – это величина, определяемая частотой сердечных сокращений и ударным объемом крови.
Ударный объем зависит от сократимости миокарда и величины показателей преднагрузки и постнагрузки. Сопротивление, против которого работает сердце, обозначается термином «постнагрузка», увеличение которой вызывает снижение ударного объема. Преднагрузка – это объем крови, который находится в полостях желудочков в конце диастолы.
При полном покое суммарная потребность в крови составляет у взрослого человека 5 л/мин/м2. При интенсивной работе она может увеличиваться в 3-6 раз. При этом сердце может перекачивать до 30 литров крови в минуту.
Большие функциональные возможности системы кровообращения обеспечиваются как внутрисердечной, так и общей нервно-гуморальной регуляцией. Это обеспечивает координированную работу сердца, его связь с сосудами и другими системами. Поэтому на повышение требований к кровообращению реагирует не только сердце или сосуды (изменение тонуса, перераспределение регионального кровообращения), но и система дыхания (увеличение легочной вентиляции), система утилизации кислорода тканями и система кроветворения (активация эритропоэза).
Сердечная недостаточность
Сердечная недостаточность - патологическое состояние, при котором развивается несоответствие между предъявляемой к сердцу нагрузкой и его способностью производить работу, которая определяется количеством притекающей к сердцу крови и сопротивлением изгнанию крови в аорте и лёгочной артерии. СН возникает в результате неспособности сердца поддерживать минутный объем кровотока, адекватный для метаболических потребностей тканей и органов. При этом уменьшение МОК не связано с уменьшением венозного возврата к сердцу. Сердечная недостаточность – это патофизиологический синдром, проявляющийся неспособностью сердца из-за ухудшения его насосной функции обеспечивать адекватное кровоснабжение организма, то есть «сердечная недостаточность – это состояние, характеризующееся снижением резервных возможностей сердца».
Это нарушение сократительной способности (насосной функции) сердца, приводящее к недостаточному снабжению клеток различных органов и тканей кислородом и питательными веществами. Основной критерий СН – снижение или неадекватность потребностям тканей минутного объёма кровотока (МОК).
Для сократительного процесса необходимы ионы кальция. Непосредственным поставщиком энергии, затрачиваемой в первый момент сокращения сердечной мышцы, являются макроэргические фосфорсодержащие соединения – аденозинтрифосфат и креатинфосфат. Ресинтез этих соединений происходит за счет энергии дыхательного и гликолитического фосфорилирования, т.е. за счет энергии, поставляемой углеводами. В сердечной мышце преобладают аэробные процессы, идущие с использованием кислорода, поэтому миокард очень чувствителен к недостатку кислорода.
Итак, работа сердца как единого насосного органа, зависит от слаженной работы мышечных волокон каждого его отдела, последовательности сокращений этих отделов, ритма и частоты сокращений сердца. Все это обеспечивается основными свойствами сердца: автоматизмом, возбудимостью, проводимостью и сократимостью.
Классификации СН
По причинам развития сердечной недостаточности:
Поражение самой мышцы сердца:
· коронарогенное;
· некоронарогенное.
Сердечную недостаточность можно рассматривать как нарушение, возникающее в результате повреждения самой мышцы сердца, и в этом случае уместен термин миокардиальная недостаточность. Она развивается при поражении мышцы сердца, например, при ИБС, инфаркте миокарда, патологии клапанов сердца, миокардитах и т.д.
Патогенетическая классификация сердечной недостаточности.
Вследствие поражения мышцы сердца – миокардиальная недостаточность:
- коронарогенная;
- некоронарогенная.
Вследствие перегрузки сердца:
- из-за увеличения преднагрузки (перегрузка объемом);
- из-за увеличения постнагрузки (перегрузка давлением – сопротивлением).
Уменьшение конечного диастолического заполнения полостей сердца.
Нарушение ритма сердца.
По течению различают:
острую (протекает несколько минут, часов или дней) и хроническую (длится годами).
По степени вовлечения в процесс отделов сердца:
· левожелудочковую (ЛЖ), характеризующуюся повышением давления в сосудах малого круга кровообращения и часто приводящую к застою в малом круге кровообращения и отеку легких;
· правожелудочковую (ПЖ), ведущую к застою в венах большого круга и расстройству функций внутренних органов;
· тотальную.
Хотя сердце является одним органом, оно анатомически и функционально может быть разделено на два отдела. При некоторых патологических процессах быстрее проявляется недостаточность одного из отделов, то есть с клинической точки зрения лево- и правожелудочковая недостаточность может встречаться изолированно. Однако, так как сосудистая система замкнута, недостаточность одного отдела не может долго существовать изолированно, она вызовет увеличение нагрузки на другой отдел, то есть неминуемо возникнет тотальная недостаточность.
Левожелудочковая недостаточность. Наиболее часто возникает при:
ишемической болезни сердца;
хронической гипертензии;
поражении аортального или митрального клапанов (при ревматизме, кальнифицированном стенозе аорты).
Обычно при ЛЖН (кроме стеноза митрального клапана) развивается дилятация левого отдела сердца (или левого предсердия). Поражаются следующие органы:
Легкие. Увеличивается давление в легочных венах и далее в легочных капиллярах. Это приводит сначала к образованию периваскулярного транссудата, затем к прогрессированию отека, вплоть до появления в альвеолярных пространствах эритроцитов. Эти патологические изменения клинически проявляются диспноэ - ранний и четкий симптом появления ЛЖН. Далее развивается ортопноэ и пароксизмальное (сердечное) ночное диспноэ. Появляется кашель.
Почки. Уменьшается перфузия почек. Активируется ренин-ангиотензин-альдостероновая система - задержка жидкости и усиление отека легких. В почках из-за гипоперфузии развивается тубулярный ишемический некроз, постепенно проявляются нарушения функций почек - появляется азотемия.
Мозг. Из-за церебральной гипоксии может развиться гипоксическая энцефалопатия в терминальных стадиях вплоть до ступора и комы.
Правожелудочковая недостаточность. Чаще сопровождает ЛЖН, так как увеличивается нагрузка на правый желудочек из-за повышения сопротивления его работе. Редко изолированно - “легочное сердце” - из-за патологии легких или легочных сосудов, что также увеличивает нагрузку на правый желудочек. Другие варианты - различные формы кардиомиопатий и диффузный миокардит, которые поражают правый желудочек чаще. Редко возникает из-за повреждения трикуспидального и легочного клапанов.
Более выражены поражения печени, почек, застойная спленомегалия. Развивается отек вплоть до анасарки. В патогенезе отека наиболее важно увеличение в плазме альдостерона из-за недостаточности функции печени (вторичный гиперальдостеронизм). Развивается портальная гипертензия и асцит. При ПЖН меньше выражены симптомы со стороны легких.
Недостаточность сердца в результате уменьшения КДО желудочков
Нарушение конечного диастолического заполнения желудочка может возникать при стенозе атриовентрикулярного отверстия, заполнении полости перикарда кровью или экссудатом (тампонада сердца), при опухоли средостения, сдавливающей сердце или вены, впадающие в сердце. При этом уменьшение МОК возникает из-за снижения систолического объема, которое не компенсируется увеличением ЧСС.
Недостаточность сердца при аритмиях
При нарушениях ритма сердца уменьшение МОК может развиваться двумя путями. При значительном уменьшении частоты сердечных сокращений (брадиаритмии) систолический объем увеличивается очень незначительно (за счет удлинения диастолы), что не может компенсировать уменьшение ЧСС.
При резком увеличении ЧСС происходит укорочение диастолы в такой степени, что ее продолжительности оказывается недостаточным для нормального диастолического заполнения. В результате снижается систолический объем. При такой форме нарушения ритма, как фибрилляция желудочков, происходит не координированное сокращение кардиомиоцитов, что приводит к резкому снижению систолического объема.
Недостаточность сердца в результате перегрузки
Как уже говорилось, недостаточность сердца в результате перегрузки может быть обусловлена либо повышением сопротивления выбросу - перегрузка давлением (или увеличение постнагрузки), либо увеличением диастолического наполнения желудочков, в этом случае говорят о перегрузке объемом (или увеличении преднагрузки),
Перегрузка объемом обусловлена увеличенным поступлением крови в полость желудочка в диастолу. Возникает в следующих случаях:
· при увеличении венозного возврата; при увеличении ОЦК;
· при недостаточности клапанов аорты и/или легочной артерии в результате регургитации крови.
Перегрузка давлением возникает при повышении сопротивления выбросу:
· при повышении давления в аорте или/и легочной артерии (гипертоническая болезнь, симптоматические гипертензии, легочные гипертензии при заболеваниях легких, тромбоэмболии легочной артерии);
· при сужении выходного отверстия из полости желудочка (стеноз устья аорты, стеноз отверстия легочного ствола);
· при коарктации аорты;
· при системном атеросклерозе.
Механизмы компенсации работы сердца при повышенной нагрузке
1) Механизм компенсации при перегрузке объемом - гетерометрический:
Зависимость силы сокращения от исходной длины мышечных волокон является решающим фактором, определяющим функцию сердечной мышцы. Она лежит в основе закона сердца Франка-Старлинга, который утверждает, что в определенных пределах увеличение конечного диастолического объема желудочка, являющегося производным от длины мышцы, приводит к усилению сокращения желудочка. Происходящее при этом расширение полостей сердца называется тоногенной дилатацией.
Сила сокращения поперечнополосатой мышцы любого типа, включая сердечную мышцу, зависит от ее исходной длины. Наиболее мощное сокращение саркомера наблюдают при длине 2.2 мкм. Именно при такой длине саркомера расположение обоих видов миофиламентов по отношению друг к другу наиболее благоприятно для их взаимодействия. Имеются данные о том, что длина саркомера определяет также степень активности контрактильной системы, зависящей от ее чувствительности к ионам Са2+. Максимальная активность наблюдается при длине саркомера 2.2 мкм. Если длина саркомера увеличивается до 3.65 мкм, то создаваемое напряжение падает до нуля, а актиновые волокна полностью выходят из зоны контакта с миозиновыми. С другой стороны, если длина саркомера менее 2.0 мкм, то происходит скручивание миозиновых нитей и их перегиб. Одновременно снижается чувствительность контрактильных локусов к ионам Са2+, а, следовательно, и сила сокращения.
2) Механизм компенсации при перегрузке давлением - гомеометрический. Длина мышцы в данном случае почти не увеличивается, а повышается сила сердечного сокращения и интенсивность, особенно в конце диастолы. Причем повышение силы сердечных сокращений происходит не сразу, а постепенно - с каждым последующим сокращением.
Тахикардия. Прямая зависимость МОК от ЧСС существует в определенных пределах. Если частота возрастает выше критического уровня (90 - 100) падает сила сокращений из-за чрезмерного накопления продуктов метаболизма. Уменьшается период диастолы - время заполнения желудочков и питания сердца по коронарам.
Симпатическая стимуляция - максимальный МОК от 170 до 220 (усиливается сила сокращений и уменьшается время систолического сокращения, что удлиняет диастолу).
Миокардиальная недостаточность
Заболевания сердца:
85 - 90 % всех заболеваний сердца приходится на 4 группы:
1) ИБС;
2) заболевания, вызываемые гипертензией и легочной гипертензией;
3) врожденные поражения;
4) нарушения клапанов (кальцинифицированный склероз аортального клапана, поражение митрального клапана, инфекционный эндокардит и ревматизм сердца).
Коронарогенные поражения мышцы сердца возникают при ишемии, причиной которой могут быть: поражение коронарных артерий (например, атеросклеротическое поражение), тромбоз коронарных артерий, эмболия и т.д., приводящая к дефициту энергии (АТФ) и снижению сердечного выброса. Главная опасность нарушения коронарного кровообращения заключается в дефиците кислорода, т.е. развитии гипоксии. Непосредственным источником энергии для сокращения сердца являются АТФ и креатинфосфат. В связи с тем, что обменные процессы сердца целиком зависят от образования энергии в результате окислительных процессов, внезапное прекращение коронарного кровотока уже через несколько минут приводит к тяжелым нарушениям деятельности сердца. Если отсутствие кислорода (аноксия) продолжается более 30 минут, то происходят необратимые структурные изменения миокарда. Восстановить деятельность сердца по истечению этого срока невозможно (предел реанимации).
ИБС
80% всей смертности от сердечной патологии приходится на ИБС. Снижение смертности - 45% из-за лечения, 55% - изменения стиля жизни. В патогенезе можно выделить 5 основных механизмов:
1) атеросклеротический стеноз коронарных артерий (уменьшает до 75 % коронарный кровоток, поражаются основные ветви);
2) агрегация тромбоцитов в коронарной системе (атеросклероз обнажает коллаген - тромбоз, медиаторы, употребление аспирина, рыбная диета с ПНЖК);
3) коронарный спазм (даже без атеросклероза, способствует тромбозу, усиление прессорной инервации медиаторами из тромбоцитов);
4) неатеросклеротическое поражение коронаров (эмболия, артерииты, травма);
5) гемодинамические изменения (шок, подъем давления в правом предсердии с регургитацией через трикуспидальный клапан снижает разницу в давлениях и уменьшает перфузию, ЛЖН).
Сердце (в отличие, например, от скелетной мускулатуры) - “всеядный” орган. Особого интереса заслуживает значительная доля свободных жирных кислот среди потребляемых сердцем веществ, а также тот факт, что сердце в отличие от скелетных мышц способно использовать лактат. Вследствие такой способности сердца потреблять все доступные вещества главная опасность нарушения коронарного кровообращения заключается не в недостатке субстрата, а в дефиците кислорода.
Метаболическое расщепление различных веществ сопровождается образованием АТФ - непосредственного источника энергии для сокращений сердца. Содержание АТФ в миокарде невелико по сравнению с тем, которое требуется для его сократительной деятельности. За несколько секунд работы сердца количество АТФ обновляется (то есть расщепляется с образованием АДФ и неорганического фосфата и вновь ресинтезируется) несколько раз. В сердце имеется еще один макроэргический фосфат - креатинфосфат, содержание которого примерно равно содержанию АТФ. Количество креатинфосфата служит особо чувствительным показателем снабжения миокарда питательными веществами и кислородом, так как его расщепление лежит в основе метаболического ресинтеза АТФ.
В связи с тем, что обменные процессы сердца почти целиком зависят от образования энергии в результате окислительных процессов, внезапное прекращение коронарного кровотока уже через несколько минут приводит к тяжелым нарушениям деятельности сердца. Грубые нарушения энергообразования в миокарде проявляются резким падением содержания макроэргических фосфатов (креатинфосфата и АТФ). Хотя сердце способно в незначительной степени осуществлять анаэробный гликолиз, в результате которого образуется лактат, однако в отсутствие кислорода лактат не утилизируется. Если аноксия продолжается больше 30 мин, происходят необратимые структурные изменения миокарда, восстановить деятельность сердца по истечению этого срока невозможно (предел реанимации).
Некоронарогенные повреждения сердца возникают при различных процессах, повреждающих миокард: инфекционные процессы в миокарде (инфекционный миокардит), иммунное повреждение, травма сердца, электролитный дисбаланс, авитаминозе и т.д. При этих повреждениях существенно возрастает «запрос» и расход миокардом кислорода и продуктов обмена в сравнении с уровнем их притока.
Патогенез миокардиальной сердечной недостаточности.
Для практических целей особенно важно выделять два типа миокардиальной недостаточности, различающихся по особенностям метаболизма макроэргов, в частности креатинфосфата.
Первый характеризуется угнетением ресинтеза креатинфосфата вследствие недостаточности выработки энергии; он наблюдается при гипоксии миокарда, действии на него метаболических ядов и пр. Снижение сократимости в этом случае наступает в результате того, что в связи с недостатком креатинфосфата уменьшаются запасы энергии, необходимой для деятельности сократительных белков. Кроме того, нехватка АТФ приводит к нарушению функционирования мембранных энергозависимых ионных насосов, что приводит к нарушению возбудимости и проводимости миокарда.
При втором типе запасы макроэргов достаточны, но они не могут быть эффективно использованы из-за нарушения электромеханического сопряжения. Этот тип наблюдается при гипокальциемии, передозировке антагонистов кальция, отравлении местными анестетиками, барбитуратами и пр. Сократимость восстанавливается при введении веществ, улучшающих электромеханическое сопряжение (катехоламинов, сердечных гликозидов). Напротив, при снижении запасов энергии такие препараты могут даже ухудшить состояние. В подобных случаях следует снижать энергозатраты сердца, облегчая его работу.
Острая сердечная недостаточность
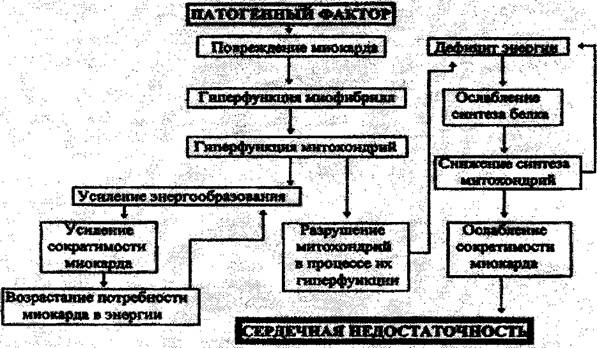
Патогенетические механизмы развития острой недостаточности сердца, возникающей при чрезмерной перегрузке миокарда, представлены на рисунке. Как следует из рисунка, развитие острой сердечной недостаточности начинается с воздействия на сердце патогенного фактора, причем в данном случае предполагается, что этот фактор по своей интенсивности является чрезвычайным. Повреждение миокарда сопровождается снижением сократительной функции сердца, что может привести к серьезным расстройствам гемодинамики. Справиться с ними сердечная мышца может только при условии значительного усиления функции миофибрилл, оставшихся неповрежденными.
Гиперфункция миофибрилл требует соответствующего энергетического обеспечения, в результате чего возникает гиперфункция митохондрий. Вырабатывающаяся в митохондриях энергия практически полностью идет на обеспечение сократительного акта, что позволяет миофибриллам в течение определенного периода работать в режиме гиперфункции. С другой стороны, гиперфункция митохондрий приводит к их усиленному разрушению, нарастанию дефицита энергии в миокарде, ослаблению синтеза белка и снижению образования митохондрий. Вследствие этого, возникает энергетическое истощение и ослабление сократимости миокарда.
Гипертрофия миокарда как компенсаторная реакция: стадии формирования
Гипертрофия миокарда – универсальный механизм компенсации перегрузки сердца.
Любая перегрузка сердца приводит к увеличению напряжения миокарда соответствующей камеры сердца. При перегрузке сопротивлением максимальное напряжение миокард испытывает во время систолы (систолическая перегрузка), при перегрузке объемом – во время диастолы (диастолическая перегрузка). Увеличение напряжения миокарда является главным фактором развития гипертрофии миокарда. При гипертрофии миокарда увеличивается объем каждого мышечного волокна, а общее количество волокон не изменяется.
Выделяют три стадии развития гипертрофии.
Аварийная стадия развивается сразу при предъявлении сердцу повышенной нагрузки.
Стадия завершившейся гипертрофии и относительно устойчивой гиперфункции.
Увеличение массы миокарда обеспечивает длительную компенсацию к условиям повышенной нагрузки.
Стадия изнашивания структур и прогрессирующего кардиосклероза. В эту стадию развивается недостаточность гипертрофированного сердца.
I стадия - аварийная. Развивается непосредственно после выполнения нагрузки. Клинически эта стадия соответствует острой сердечной недостаточности. У больного отмечается резкое падение кровяного давления, нитевидный пульс, холодный пот, возможна потеря сознания, то есть все клинические признаки гемодинамической катастрофы. Характеризуется сочетанием патологических изменений в миокарде (исчезновение гликогена, снижение уровня креатинфосфата, уменьшение содержания внутриклеточного калия и повышение содержания натрия, мобилизация гликолиза, накопление лактата) с мобилизацией резервов миокарда и организма в целом. В этой стадии повышены нагрузка на единицу мышечной массы и интенсивность функционирования структуры (ИФС); происходит быстрое, в течение недель, увеличение массы сердца вследствие усиленного синтеза белков и утолщения мышечных волокон. В эту стадию в миокарде возникают очаги дистрофии и некроза, падает содержание АТФ и КрФ, повышается уровень АДФ, АМФ и аденозина. За счет повышения интенсивности функционирования структур увеличивается продукция метаболитов изнашивания и достаточно быстро формируется патологическая гипертрофия миокарда. Стимулируется анаэробный метаболизм и развивается внутриклеточный ацидоз. В части кардиомиоцитов ионы водорода вызывают блокаду активных участков в молекуле тропонина и не позволяют ионам кальция снять тропомиозиновый блок с актина. Эти мышечные волокна находятся в состоянии расслабления и не могут сокращаться при приходе ПД, то есть в миокарде возникают участки асистолии. В другой части кардиомиоцитов на первый план выходит дефицит энергии и нарушение функционирования Са+-насосов, обеспечивающих расслабление кардиомиоцитов. При выраженном дефекте диастолы эти клетки могут находиться в состоянии контрактуры -- стойкого сокращения без расслабления. Мозаичное распределение в миокарде участков асистолии и контрактуры ведет к уменьшению общей сократимости миокарда и, следовательно, насосной функции сердца, что ведет к снижению ударного и минутного сердечного выброса и является непосредственной причиной острой сердечной недостаточности.
В аварийную стадию летальность достигает 20%. При достаточности компенсаторных возможностей сердечно-сосудистой системы процесс переходит во вторую фазу.
II стадия -- относительной устойчивости (завершившейся патологической гипертрофии миокарда). Процесс гипертрофии завершен, масса миокарда увеличена на 100-120% и больше не прибавляется, ИФС нормализовалась. В эту фазу в миокарде уменьшаются очаги дистрофии и некроза, восстанавливается метаболизм, нормализуется содержание макроэргов, снижается ацидоз. За счет сформировавшейся гипертрофии нагрузка распределяется в большей массе миокарда и снижается нагрузка на единицу массы. Нормализовались гемодинамические показатели. Гипертрофированное сердце приспособилось к новым условиям нагрузки и в течение длительного времени компенсирует ее. Поскольку механизмы адаптации в эту фазу работают с большим напряжением, больному противопоказаны повышенные нагрузки, что может провоцировать повторение аварийной фазы. Постепенно эта фаза переходит в третью стадию.
III стадия -- стадия постепенного истощения и прогрессирующего кардиосклероза. Характеризуется глубокими обменными и структурными изменениями, которые исподволь накапливаются в энергообразующих и сократительных элементах клеток миокарда. Сохраняющиеся в миокарде мелкие очаги дистрофии и некроза являются источниками аутоантигенов, против которых начинают вырабатываться аутоантитела и цитотоксические CD8+ Т-лимфоциты. Под влиянием лимфокинов и монокинов активируется пролиферация фибробластов и продукция ими коллагена. В миокарде развивается соединительная ткань, что в сочетании с повреждением кардиомиоцитов ведет к постепенному снижению сократительной способности миокарда. ИФС снова возрастает. Нарушается регуляторный аппарат сердца. Прогрессирующее истощение компенсаторных резервов приводит к возникновению хронической недостаточности сердца, а в дальнейшем - к недостаточности кровообращения.
Повышенная нагрузка на организм приводит к усиленной функции сердца. При этом активируется работа всех его элементов (клеток, внутриклеточных органоидов). Включаются все механизмы адаптации сердца при перегрузке. Повышается интенсивность функционирования структур. При этом в клетке и в органе происходит накопление метаболитов изнашивания - группы молекул, образующихся в интенсивно работающих клетках. К метаболитам изнашивания относятся: молочная, пировиноградная кислоты, углекислый газ, ионы водорода, продукты распада АТФ, креатин, оксид азота и др. Под влиянием метаболитов изнашивания происходит активация кислых ядерных белков, которые снимают гистонный блок с ДНК, активируя транскрипцию ряда структурных генов. Усиливается синтез иРНК, которая поступает на рибосомы и участвует в биосинтезе новых клеточных белков.
Увеличение массы сердца происходит вследствие утолщения каждого мышечного волокна, что сопровождается изменением соотношения внутриклеточных структур. Объем клетки при этом увеличивается пропорционально кубу линейных размеров, а поверхность - пропорционально их квадрату, что приводит к уменьшению клеточной поверхности и на единицу массы клетки. Известно, что через поверхность клетки происходит ее обмен с внеклеточной жидкостью - поглощение кислорода, питательных веществ, выведение продуктов метаболизма, обмен воды и электролитов. В силу перечисленных изменений возникают условия для ухудшения снабжения мышечного волокна, особенно его центральных отделов.
Гипертрофированное сердце отличается от нормального по ряду обменных, функциональных и структурных признаков, которые, с одной стороны, позволяют ему длительное время преодолевать повышенную нагрузку, а с другой – создают предпосылки для возникновения патологических изменений. В гипертрофированном миокарде ухудшается энергетическое, сосудистое, регуляторное обеспечение, уменьшается функциональный резерв, развиваются дистрофические изменения в кардиомиоцитах и постепенно наступает их гибель. Замещение кардиомиоцитов соединительной тканью приводит к углублению обменных нарушений и развитию дистрофических изменений.
Дистрофические изменения сердечной мышцы сопровождаются расширением полостей сердца, снижением силы сердечных сокращений – возникает миогенная дилатация сердца, сопровождающаяся увеличением остающейся крови в полостях сердца и переполнением вен. Увеличение давления крови в полостях правого предсердия и в полых венах приводят к тахикардии, что усугубляет обменные нарушения в миокарде. Поэтому расширение полостей сердца и тахикардия являются грозным симптомом начинающейся декомпенсации сердца.
Морфофункциональные особенности гипертрофированного миокарда, приводящие
к сердечной декомпенсации
Клеточная мембрана играет большую роль в проведении возбуждения и в сопряжении процессов возбуждения и сокращения, осуществляемом через тубулярную систему и саркоплазматический ретикулум. Поскольку рост этих образований при гипертрофии мышечного волокна также отстает, то создаются предпосылки для нарушения процессов сокращения и расслабления кардиомиоцитов: вследствие замедления выхода ионов кальция в миоплазму ухудшается сокращение, а в результате затруднения обратного транспорта ионов кальция в ретикулум - расслабление, иногда могут возникать локальные контрактуры отдельных кардиомиоцитов.
При гипертрофии увеличение объема клетки происходит в большей степени, чем объема ядра. Способность ядра высокодифференцированной клетки к делению резко ограничено. При этом увеличиваются только линейные размеры ядер за счет увеличения числа хромосом, что сопровождается некоторым увеличением содержания ДНК. А так как роль ядра заключается в обеспечении белкового синтеза, а, следовательно, и процессов восстановления внутриклеточных структур, то относительное уменьшение ядра может привести к нарушению синтеза белков и ухудшению пластического обеспечения клетки.
В процессе развития гипертрофии масса митохондрий вначале увеличивается быстрее, чем масса сократительных белков, создавая условия для достаточного энергетического обеспечения и хорошей компенсации функции сердца. Однако в дальнейшем, по мере усугубления процесса, увеличение массы митохондрий начинает отставать от роста массы цитоплазмы. Митохондрии начинают работать с предельной нагрузкой, в них развиваются деструктивные изменения, снижается эффективность их работы, нарушается окислительное фосфорилирование. Это ведет к ухудшению энергетического обеспечения гипертрофированной клетки.
Увеличение массы мышечных волокон зачастую не сопровождается адекватным увеличением капиллярной сети, особенно в случаях быстрого развития недостаточности сердца. Крупные венечные артерии также не обладают необходимым приспособительным ростом. Поэтому во время нагрузки ухудшается сосудистое обеспечение гипертрофированного миокарда. В гипертрофированном сердце нарушена структура вставочных дисков и Z-полос, что имеет своим следствием изменение электрической активности миокарда, ухудшение координированности сокращения сердца в целом.
При развитии гипертрофии миокарда в процесс обязательно вовлекается нервный аппарат сердца. Наблюдается усиленное функционирование внутрисердечных и экстракардиальных нервных элементов. Однако рост нервных окончаний отстает от роста массы сократительного миокарда. Происходит истощение нервных клеток; нарушаются трофические влияния, уменьшается содержание норадреналина в миокарде, что ведет к ухудшению его сократительных свойств, затруднению мобилизации его резервов. Следовательно, нарушается и регуляторное обеспечение сердца.
Гипертрофированное сердце вследствие увеличения массы его сократительного и энергообеспечивающего аппарата способно длительное время выполнять значительно большую работу, чем сердце нормальное, сохраняя при этом нормальный метаболизм. Однако способность приспосабливаться к изменяющейся нагрузке, диапазон адаптационных возможностей у гипертрофированного сердца ограничены. Уменьшен функциональный резерв. Это делает гипертрофированное сердце в силу указанной выше несбалансированности внутриклеточных и тканевых структур более ранимыми при различных неблагоприятных обстоятельствах.
Длительная и интенсивная нагрузка на сердечное мышечное волокно ведет к его истощению и нарушению функций. При этом могут возникнуть нарушения сократительной функции мышечного волокна вследствие нарушения образования энергии митохондриями и нарушения использования энергии сократительным аппаратом. При разных формах недостаточности сердца один из этих патологических вариантов может преобладать, в частности при длительной гиперфункции сердца ведущим является нарушение использования энергии. При этом наряду с плохой сократимостью наблюдается затруднение расслабления мышечного волокна, возникновение мышечных локальных контрактур, а в дальнейшем - дистрофия и гибель кардиомиоцитов.
Повышенная нагрузка неравномерно распределяется между различными группами мышечных волокон: более интенсивно функционирующие волокна быстрее выходят из строя, гибнут и замещаются соединительной тканью (кардиосклероз), а оставшиеся принимают на себя все более повышенную нагрузку. Кардиосклероз ведет к сдавлению кардиомиоцитов, изменению механических свойств сердца, еще большему ухудшению диффузии, углублению обменных нарушений. Считается, что при замене соединительной тканью 20-30% массы сердца его нормальная работа невозможна.
Дистрофические изменения сердечной мышцы сопровождаются расширением полостей сердца, снижением силы сердечных сокращений - возникает миогенная дилатация сердца, сопровождающаяся увеличением остающейся во время систолы в полостях сердца крови и переполнением вен. Повышенное давление крови в полостях правого предсердия и отверстиях полых вен прямым действием на синуснопредсердный узел и рефлекторно (рефлекс Бейнбриджа) вызывает тахикардию, которая усугубляет обменные нарушения в миокарде. Поэтому расширение полостей сердца и тахикардия служат грозными симптомами начинающейся декомпенсации.
При оценке биологического значения гипертрофии миокарда следует обратить внимание на внутреннюю противоречивость данного явления. С одной стороны, это весьма совершенный приспособительный механизм, который обеспечивает длительное выполнение сердцем повышенной работы в нормальных и патологических условиях, а с другой - особенности структуры и функции гипертрофированного сердца служат предпосылкой для развития патологии. Преобладание одной из сторон в каждом конкретном случае определяет особенности протекания патологического процесса.
Виды гипертрофии миокарда
Различают физиологическую и патологическую гипертрофию миокарда. Физиологическая гипертрофия возникает при действии на сердце значительных периодических нагрузок с паузами. Такая гипертрофия развивается, например, в сердце спортсмена, у которого периоды тренировок чередуются с периодами отдыха. Физиологическая гипертрофия формируется медленно, постепенно. За ростом мышечных волокон успевают расти капилляры и нервы, т.е. сохраняются нормальное кровоснабжение и нервная трофика. В миокарде повышается количество митохондрий, возрастает активность ферментов дыхательной цепи и АТФазная активность головок миозина, увеличивается содержание миоглобина, повышается мощность мембранных ионных насосов, сохраняется нормальное отношение объема ядра к объему цитоплазмы (1:5). Физиологическая гипертрофия - это сбалансированный рост кардиомиоцитов, позволяющий им эффективно справляться с возросшей нагрузкой.
Патологическая гипертрофия формируется при действии на сердце постоянной большой нагрузки без пауз (например, при пороке сердца). Гипертрофия в этом случае развивается очень быстро. За ростом мышечных волокон не успевают расти кровеносные капилляры и нервы, нарушается доставка кислорода к кардиомиоцитам и нервная трофика. Уменьшается количество митохондрий и других клеточных органелл, приходящихся на единицу объема клетки. Снижается мощность мембранных ионных насосов, обеспечивающих реполяризацию мембраны кардиомиоцитов и удаление из миоплазмы избытка ионов Са2+. Возрастает ядерно-цитоплазматический индекс (до 1:15), что создает препятствие для диффузии кислорода к ядру. Патологическая гипертрофия - это несбалансированный рост, позволяющий, однако, сердцу на какое-то время адаптироваться к повышенной нагрузке. С течением времени патологическая гипертрофия приводит к изнашиванию миокардиальных структур. Этому способствует и возникающее ремоделирование миокарда, под которым понимается изменение архитектоники сердечной мышцы, вследствие нарушения пространственного соотношения между кардиомиоцитами, кровеносными и лимфатическими капиллярами, нервными волокнами и соединительными элементами, приводящее к изменению геометрии камер сердца. Нарушение оптимальной структуры миокарда в конечном итоге приводит к нарушению его систолической и диастолической функции.
Хроническая сердечная недостаточность
Хроническая – застойная недостаточность развивается постепенно. При этом из-за недостаточности выброса крови из сердца уменьшается кровенаполнение органов на путях оттока. Одновременно из-за неспособности сердца перекачать всю притекающую кровь развивается застой и на путях притока.
При левожелудочковой недостаточности застой крови наблюдается в венах малого круга, что может привести к отеку легких. При правожелудочковой недостаточности застой наблюдается в венах большого круга кровообращения, при этом увеличивается печень, появляются отеки на ногах, асцит (скопление жидкости в брюшной полости).
При хронической недостаточности снижается минутный объем крови, замедляется скорость кровотока, повышается венозное давление, капиллярные сосуды и посткапиллярные вены расширены, кровоток в них замедлен, давление повышено. Замедление кровотока в большом круге кровообращения и нарушение кровообращения в легких приводит к увеличению в крови восстановленного гемоглобина. Это придает коже и слизистым синюшный оттенок – цианоз. Тканям не хватает кислорода.
Гипоксия приводит к накоплению недоокисленных продуктов обмена и углекислоты – развивается ацидоз. Ацидоз и гипоксия ведут к нарушению регуляции дыхания, возникает одышка. С целью компенсации гипоксии стимулируется эритропоэз, увеличивается общий объем крови и относительное содержание клеток в крови, что способствует повышению вязкости крови и ухудшению ее гемодинамических свойств.
Повышение давления в венозных капиллярах и ацидоз приводят к развитию отека, который усиливает гипоксию. Развитию застойного отека способствует общее нарушение водно-электролитного обмена, сопровождающееся задержкой в организме натрия и воды.
Длительная недостаточность кровообращения из-за циркуляторной гипоксии ведет к глубокому и необратимому изменению внутриклеточного метаболизма. Это сопровождается нарушением синтеза белков, в том числе и белков дыхательных ферментов, что приводит к развитию гистотоксической гипоксии. Эти симптомы характерны для терминальной фазы недостаточности кровообращения. При прогрессировании недостаточности кровообращения наступает тяжелое истощение – сердечная кахексия.
Основными проявлениями недостаточности являются:
Повышение конечного диастолического давления в полостях сердца.
В норме после каждой систолы не происходит полного опорожнения полостей сердца. Объем оставшейся в полостях сердца крови называется конечным диастолическим объемом. При недостаточности сократительной функции миокарда этот объем увеличивается (увеличение преднагрузки). Это приводит к повышению давления в полостях. В норме это давление равно 5-10 мм рт. ст. При левожелудочковой недостаточности оно увеличивается до 20-30 мм рт. ст., при правожелудочковой недостаточности до 30-40 мм рт. ст. Это увеличивает нагрузку на миокард из-за увеличения преднагрузки.
Дилятация полостей (расширение полостей).
Если дилатация развивается на ранних стадиях перегрузочной формы недостаточности (тоногенная дилатация), то она имеет саногенетический характер, т.к. способствует увеличению силы сердечного сокращения, что позволяет преодолеть повышенную нагрузку. Если дилатация миогенная, то это плохой прогностический признак, свидетельствующий о нарушении сократительной способности миокарда.
Уменьшение минутного объема сердца (количество крови, выбрасываемое желудочками за 1 минуту в покое равно в норме 5,5 литрам) сопровождается падением артериального давления и повышением венозного давления.
Механизмы развития сердечных отеков
1. Повышение венозного давления приводит к нарушению оттока лимфы и усилению фильтрации плазмы из сосудов в ткань. В «застойной» печени уменьшается выработка белков (альбуминов) – развивается гипоонкия плазмы. Это усиливает фильтрацию жидкости из сосудов в ткань.
2. Падение артериального давления уменьшает кровоток в почках. Это стимулирует выработку ренина, активируется секреция альдостерона, усиливается реабсорбция Na+ и воды.
3. Усиленная реабсорбция Na+ приводит к гиперосмии. Раздражение осморецепторов рефлекторно усиливает секрецию антидиуретического гормона, который увеличивает реабсорбцию воды, способствуя ее задержке в тканях.
4. Уменьшение минутного объема, падение артериального давления, повышение венозного давления приводят к развитию циркуляторной гипоксии. Повышается проницаемость стенки сосудов, усиливается фильтрация жидкой части крови в ткань, что способствует развитию отека.
Хроническая сердечно-легочная недостаточность
«Легочное сердце» или сердечно-легочная недостаточность обусловлена повышением сопротивления сердечному выбросу (увеличение постнагрузки) в сосудах малого круга, (массивный тромбоз, эмболия сосудов малого круга кровообращения), что приводит к декомпенсации правого желудочка. Эта недостаточность может быть острой и хронической. При этом на первый план выступают симптомы перегрузки правого желудочка и относительной коронарной недостаточности этого отдела сердца.
Хроническая сердечно-легочная недостаточность возникает при постепенном нарастании сопротивления в сосудах малого круга, что наблюдается при пневмосклерозе, эмфиземе легких, первичной артериальной гипертонии малого круга кровообращения. При хронической сердечно-легочной недостаточности наблюдается прогрессивно развивающаяся декомпенсация сердечной деятельности по правожелудочковому типу. Прогноз плохой.
Застой венозной крови во внутренних органах из-за развития тканевой гипоксии приводит к различным дистрофическим проявлениям и разрастанию межуточной ткани. Наблюдается уплотнение почек, селезенки, развивается сердечный цирроз печени. При застое в легких происходит диапедез (выход) эритроцитов, в альвеолы и в перегородки между ними. Макрофаги легких их фагоцитируют, в результате внутри фагоцитов образуются глыбки пигмента гемосидерина. Развитие сердечно-легочной недостаточности нередко сопровождается кровохарканьем. При застое в легких нарушается их вентиляция, что может стать причиной развития гипостатического воспаления легких.
На почве венозного застоя развиваются отеки и водянки. Застой венозной крови в слизистых оболочках дыхательных и пищеварительных путей вызывает развитие в них хронических катаральных явлений (застойные катары бронхов, желудка, двенадцатиперстной кишки и др.).
Возникающий ацидоз обусловливает раздражение дыхательного центра, что приводит к появлению мучительной одышки особенно по ночам. Этот симптом называется сердечной астмой.
Механизмы прогрессирования хронической сердечной недостаточности (роль нейрогормональных нарушений, миокардиальной и эндотелиальной дисфункции)
Эндотелий играет основную роль в регуляции тонуса сосудов и гемостаза. Все основные факторы риска развития ХСН, такие как дислипидемия, артериальная гипертония, сахарный диабет, курение и другие во многом реализуют свое патологическое влияние именно через эндотелиальную дисфункцию (ЭД). ЭД определяет развитие пролиферативных процессов в миокарде с преобладанием фиброза, отсутствие резерва дилатации микрососудистого русла и, как следствие, падение сократительной способности миокарда. При постинфарктном ремоделировании сердца отсутствие защитной роли сосудистого эндотелия приводит к быстрому развитию клинически выраженной сердечной недостаточности и в дальнейшем к смерти.
В последние годы признана важная роль эндотелиальной дисфункции в развитии и прогрессировании ХСН. В литературе описана взаимосвязь между концентрацией цитокинов в плазме крови (ФНО-α, ИЛ-1β, ИЛ-6 и ИЛ-8), вазорегулирующей и антитромбогенной активностью сосудистой стенки у больных ХСН. Изучена динамика нарушений функциональной активности эндотелия у больных ХСН различных функциональных классов тяжести. В силу крайнего разнообразия возможных последствий эндотелиальной дисфункции клинические её проявления могут быть так же крайне разнообразны.
Установлена взаимосвязь вазорегулирующей функции эндотелия и уровня цитокинов у больных ХСН с основными морфофункциональными параметрами сердца. Выявлена связь между антитромбогенной, вазорегулирующей активностями эндотелия сосудистой стенки и уровнями провоспалительных цитокинов у больных ХСН различного класса тяжести и их взаимосвязь с клиническими проявлениями заболевания. Отмечены взаимосвязи между выраженностью нарушений вазорегулирующей и антитромбогенной функции эндотелия и концентрацией провоспалительных цитокинов в плазме крови. По мере нарастания уровней провоспалительных цитокинов в плазме крови отмечено снижение эндотелийзависимой дилатации плечевой артерии и уменьшение антитромбогенного потенциала сосудистой стенки. При улучшении клинического состояния выявлена положительная динамика вазорегулирующей и антитромбогенной функции эндотелия сосудов.
Клинические и экспериментальные данные указывают на возможность восстановления нарушенной вазорегулирующей функции эндотелия и антитромбогенного потенциала сосудистой стенки под влиянием терапии статинами.













Материалы на данной страницы взяты из открытых источников либо размещены пользователем в соответствии с договором-офертой сайта. Вы можете сообщить о нарушении.
