Поделиться

ЛЕКЦИЯ
НАРУШЕНИЕ ЭНДОКРИННОЙ ФУНКЦИИ ПОДЖЕЛУДОЧНОЙ ЖЕЛЕЗЫ
Понятие об эндокринной функции поджелудочной железы
В 1869 г. П. Лангерганс впервые описал морфологический субстрат эндокринной функции pancreas, который представлен скоплениями α- (А-), β- (В-), δ- и PP-клеток. Комплекс всех выше перечисленных клеточных элементов, включая сосудистую и нервную системы, получил впоследствии наименование островков Лангерганса. В поджелудочной железе человека таких островков насчитывается около 1 млн. с общей массой 1-1,5 г (0,9-3,6 % массы железы) и размером 100-200 мкм.
В 1909 г. Миннер назвал активное вещество в экстракте pancreas инсулином. Инсулин синтезируется и секретируется β-клетками островков Лангерганса поджелудочной железы (ПЖЖ). Кроме того, островки Лангерганса секретируют глюкагон (α-клетки), соматостатин (δ-клетки) и панкреатический полипептид (РР-клетки). Здесь же встречаются энтерохромаффинные клетки D, вырабатывающие вазоактивный интестинальный полипептид (ВИП) и серотонин.
Гормоны островковых клеток взаимодействуют между собой: глюкагон в норме стимулирует секрецию инсулина, а соматостатин подавляет секрецию инсулина и глюкагона. Синтез инсулина начинается с образования препроинсулина, который расщепляется протеазой с образованием проинсулина. В секреторных гранулах аппарата Гольджи проинсулин расщепляется на инсулин и С-пептид, которые высвобождаются в кровь в процессе экзоцитоза.
Основным стимулятором секреции инсулина является глюкоза. Высвобождение инсулина в ответ на повышение уровня глюкозы в крови происходит двухфазно. Первая, или острая, фаза длится несколько минут, и она связана с высвобождением накопившегося в β-клетке инсулина в период между приемами пищи. Вторая фаза продолжается до тех пор, пока уровень гликемии не достигнет нормального тощакового (3,3-5,5 ммоль/л).
По портальной системе инсулин достигает печени - своего главного органа-мишени. Печеночные рецепторы связывают половину секретированного гормона. Другая половина, попадая в системный кровоток, достигает мышц и жировой ткани. Большая часть инсулина (80%) подвергается протеолитическому распаду в печени, остальная - в почках, и лишь незначительное количество метаболизируется непосредственно мышечными и жировыми клетками.
Основными физиологическими эффектами инсулина являются стимуляция переноса глюкозы через мембраны клеток инсулинзависимых тканей. Основными органами-мишенями инсулина являются печень, жировая ткань и мышцы. К инсулиннезависимым тканям, поступление глюкозы в которые не зависит от эффектов инсулина, в первую очередь относятся центральная и периферическая нервная система, эндотелий сосудов, клетки крови и др. Инсулин стимулирует синтез гликогена в печени и мышцах, синтез жиров в печени и жировой ткани, синтез белков в печени, мышцах и других органах. Все эти изменения направлены на утилизацию глюкозы, что приводит к снижению ее уровня в крови. Физиологическим антагонистом инсулина является глюкагон, который стимулирует мобилизацию гликогена и жиров из депо; в норме уровень глюкагона меняется реципрокно продукции инсулина.
Биологические эффекты инсулина опосредованы его рецепторами, которые расположены на клетках-мишенях. Рецептор инсулина представляет собой гликопротеин, состоящий из четырех субъединиц. При высоком уровне инсулина в крови число его рецепторов по принципу понижающей регуляции снижается, что сопровождается снижением чувствительности клетки к инсулину. После связывания инсулина с клеточным рецептором образовавшийся комплекс поступает внутрь клетки. Далее внутри мышечной и жировой клетки инсулин вызывает мобилизацию внутриклеточных везикул, которые содержат транспортер глюкозы GLUT-4. В результате этого везикулы перемещаются к клеточной поверхности, где GLUT-4 выполняет функцию входного отверстия для глюкозы. Аналогичное действие на GLUT-4 оказывает физическая нагрузка.
В организме инсулин оказывает влияние на основные виды обмена – углеводный, белковый, жировой и водно-электролитный.
I. В отношении углеводного обмена наблюдаются следующие эффекты инсулина:
· Активирует фермент гексокиназу (глюкокиназу), запуская ключевую биохимическую реакцию аэробного и анаэробного расщепления углеводов – фосфорилирование глюкозы;
· Активирует фосфофруктокиназу, обеспечивая фосфорилирование фруктозо-6-фосфата. Эта реакция, как известно, играет важную роль в процессах гликолиза и глюконеогенеза.
· Активирует гликогенсинтетазу, стимулируя синтез гликогена из глюкозы в реакциях гликогенеза.
· Тормозит активность фосфоэнолпируваткарбоксикиназу, угнетая ключевую реакцию глюконеогенеза, т.е. превращение пирувата в фосфоэнолпируват.
· Активирует синтез уксусной кислоты из лимонной в цикле Кребса.
· Облегчает транспорт глюкозы (и других веществ) через цитоплазматические мембраны, особенно в инсулин-зависимых тканях – жировой, мышечной, печеночной.
II. Роль инсулина в регуляции жирового обмена.
· Активирует фосфодиэстеразу, усиливая распад цАМФ, что вызывает торможение липолиза в жировой ткани.
· Стимулирует синтез из жирных кислот ацил-коэнзима-А, ускоряя утилизацию клетками кетоновых тел.
III. Роль инсулина в регуляции белкового обмена:
· Усиливает поглощение аминокислот.
· Стимулирует синтез белка клетками.
· Тормозит распад белка.
· Подавляет окисление аминокислот.
IV. Роль инсулина в регуляции водно-электролитного обмена:
· Усиливает поглощение мышцами и печенью калия.
· Снижает экскрецию натрия мочой.
· Способствует задержке воды в организме.
Действие инсулина на клетки-мишени инсулин-завимых тканей начинается с его соединения со специфическим гликопротеиновым рецептором. На цитоплазматических мембранах клеток этих тканей насчитывается 50000-250000 рецепторов, хотя реально функционирует лишь около 10 %. В результате взаимодействия инсулина и рецептора развиваются следующие события:
· Происходят конформационные изменения рецептора;
· Несколько рецепторов связываются между собой и образуют микроагрегат;
· Микроагрегат поглощается клеткой (интернализация рецептора);
· Формируется один или несколько внутриклеточных сигналов.
При некоторых условиях, сопровождающихся, к примеру, повышением содержания инсулина в крови, количество поверхностных рецепторов клеток-мишеней к инсулину уменьшается, и клетки становятся менее чувствительными к инсулину. Такое уменьшение количества рецепторов и снижение их чуствительности к инсулину объясняет феномен инсулинорезистентности (например, при ожирении и ИНСД, см. ниже).
Секреция инсулина стимулируется многими метаболитами и БАВ: глюкозой, маннозой, аминокислотами, особенно лейцином и аргинином, бомбезином, гастрином, панкреазимином, секретином, глюкокортикоидами, глюкагоном, СТГ, β-адреностимуляторами. Тормозят выработку инсулина гипогликемия, соматостатин, никотиновая кислота, α-адреностимуляторы. Здесь же отметим, что инсулиновая активность изменяется под влиянием содержащихся в плазме крови антагонистов инсулина, связанных с альбумином (синальбумин), β-липопротеидами и глобулинами (γ-глобулин).
Второй гормон поджелудочной железы – глюкагон представляет собой одноцепочечный полипептид, состоящий из 29 аминокислотных остатков с молекулярной массой около 3500 D. В чистом виде глюкагон был выделен в 1951 году Геде. Его содержание в крови здоровых людей натощак близко к 75-150 нг/л (активны лишь 40 % гормона). На протяжении суток он непрерывно синтезируется α-клетками островков Лангерганса. Секреция глюкагона тормозится глюкозой и соматостатином. Как указывалось, глюкагон стимулирует липолиз, кетогенез, гликогенолиз, глюконеогенез, что ведет к повышению содержания глюкозы в крови. Существенное значение в регуляции гликемии имеет его стимулирующее действие на секрецию инсулина – косвенная стимуляция через гипергликемию и быстрая прямая гетероклеточная стимуляция в пределах островка. Гормон разрушается в почках.
Механизм действия глюкагона сводится к активации через специфические рецепторы цитоплазматических мембран аденилатциклазы главным образом печени и последующего повышения содержания цАМФ в клетках. Это и приводит к гликогенолизу, глюконеогенезу и, соответственно, к гипергликемии, липолизу, кетогенезу и некоторым другим эффектам.
Сахарный диабет: понятие, классификация.
Сахарный диабет (СД) - группа обменных заболеваний, характеризующихся гипергликемией вследствие нарушения секреции и/или эффективности действия инсулина. Сахарный диабет - это целый ряд различных патологий, которые объединяет один параметр - хроническая гипергликемия. Хроническая гипергликемия, развивающаяся при СД, сопровождается развитием осложнений со стороны многих органов и систем, в первую очередь, со стороны сердца, кровеносных сосудов, глаз, почек и нервов. СД в общей сложности страдают 5-6% населения. В экономически развитых странах мира каждые 10-15 лет число больных СД возрастает в 2 раза. Ожидаемая продолжительность жизни при СД снижается на 10-15%.
Сахарный диабет - заболевание, которое характеризуется нарушением всех видов метаболизма и расстройством жизнедеятельности организма; развивается в результате гипоинсулинизма (т.е. абсолютной или относительной инсулиновой недостаточности).
Непосредственные последствия гипергликемии:
· усиленный распад липидов в клетках;
· снижение pH крови;
· накопление в крови кетоновых тел;
· выделение глюкозы с мочой;
· избыточная потеря жидкости с мочой из-за осмотического диуреза;
· обезвоживание;
· изменение электролитного состава крови;
· гликозилирование (повреждение) белков сосудистой стенки и других тканей.
Клинические признаки гипергликемии:
· сильная жажда;
· постоянное чувство голода;
· учащенное обильное мочеиспускание;
· зуд и сухость кожи;
· сильная слабость.
Классификация
Первичный сахарный диабет I типа [синонимы: инсулинозависимый, гипоинсулинемический, юношеский (ювенильный) ИЗСД)] составляет 20% от общего числа случаев первичного сахарного диабета. Подтипы: Iа – обусловлен комбинацией генетического и средового воздействия; Ib – первичный, генетически обусловленный без экзогенной провокации; Iс – с первичным поражением b-клеток экзогенными химическими и вирусными диабетогенами.
Первичный СД II типа (инсулинонезависимый, гиперинсулинемический, взрослых, пожилого возраста, тучных, ИНСД) составляет 80% всех случаев больных СД со следующими подтипами:
· IIа – ИНСД у нетучных больных;
· IIb – ИНСД у тучных больных;
· IIс – ИНСД юношеского возраста.
Термины «ИЗСД», «ИНСД» описывают особенности клинического течения (склонный к кетоацидозу и резистентный к кетоацидозу, а термины «I и II типы» относят к патогенетическим механизмам болезни (результат доминирования аутоиммунного или иных механизмов).
Вторичный СД (это гипергликемические, или диабетические синдромы, которые являются следствием болезней, поражающих поджелудочную железу или систему регуляции углеводного метаболизма).
Вторичный диабет вызванный неаутоиммунной деструкцией b-клеток (хронический панкреатит, рак, гемохроматоз, кистоз, травмы);
· вторичный диабет, вызванный эндокринными расстройствами с гиперпродукцией контринсулярных гормонов (синдром Кушинга, акромегалия, феохромоцитома, глюкагонома, гипертиреоидизм, гиперплазия эпифиза);
· вторичный ятрогенный диабет в результате применения медикаментов (кортикостероиды, АКТГ, оральные контрацептивы, пропранолол, антидепрессанты, некоторые мочегонные);
· вторичный диабет при генетически детерминированных синдромах (липодистрофии, гипоталамические формы вторичного ожирения, гликогеноз I типа, болезни Дауна, Шерешевского, Клайнфельтера.
Критерии отличия ИЗСД и ИНСД

Сахарный диабет первого типа, понятие, этиология, патогенез, последствия
Сахарный диабет (diabetes mellitus) – это заболевание, которое обусловлено абсолютным или относительным дефицитом инсулина и характеризуется нарушением вследствие этого всех видов метаболизма, и, в первую очередь, обмена углеводов. Слово «диабет» – с греч. diabetes – «прохожу через чего-нибудь», «протекаю»; слово «mellitus» – от латинского слова «мед», указывая на сладкий вкус мочи при диабете.
Основными проявлениями сахарного диабета являются следующие:
· гипергликемия (уровень глюкозы в крови выше 6,66 ммоль/л),
· глюкозурия (содержание глюкозы в моче может достигать 555-666 ммоль/л, за сутки в первичную мочу здоровых людей фильтруется до 150 г глюкозы, больных сахарным диабетом – около 300-600 г, а возможная потеря глюкозы мочой достигает 300 г/сутки),
· полиурия (суточный диурез выше 2 л, но может достигать и 12 л),
· полидипсия – (прием жидкости более 2 л в сутки), жажда,
· гиперлактацидемия (содержание лактата в крови более 0,8 ммоль/л, чаще 1,1-1,4 ммоль/л),
· гиперкетонемия – повышенное содержание в крови кетоновых тел (чаще выше 520 мкмоль/л), кетонурия,
· липемия (повышенное содержание в крови липидов, чаще выше 8 г/л),
· быстрое похудание, свойственное больным с ИЗСД.
· понижение толерантности организма к глюкозе, определяемой с помощью нагрузочной пробы глюкозой [75 г глюкозы и стакан воды, далее наличие двукратного превышения содержания глюкозы (до 11,1 ммоль/л) на протяжении 60-ой, 90-ой и 120-ой минутах определения].
Этиология. ИЗСД рассматривается как мультифакториальное наследование. Экзогенные и эндогенные факторы, вызывающие ИЗСД, стали называть диабетогенами. Диабетогенные факторы – это события, любое из которых, с определенной долей вероятности, может запустить развитие ИЗСД у носителей генетических особенностей. Вирусные и химические диабетогены способны спровоцировать аутоиммунный цитолиз b-клеток в организме генетически предрасположенных индивидов с наследственными особенностями регуляции иммунного ответа. Провоцирующее воздействие имеет наиболее важное значение в течение раннего и сравнительно ограниченного периода онтогенеза. Именно поэтому больные ИЗСД заболевают в молодом возрасте.
Как указывалось, в настоящее время говорят об инфекционных и неинфекционных диабетогенах. Среди первых фигурируют многочисленные типы вирусов: краснухи, эпидемического паротита осповакцины, Эпштейна-Барр, энтеровирус Coxsackie B4 и не Coxsackie, реовирусы, цитомегаловирусы, которые на клиническом материале и экспериментальных моделях способны спровоцировать повреждения b-клеток панкреатических островков. К примеру, до 40 % детей, рожденных от матерей, перенесших в третьем триместре краснуху, заболевают ИЗСД в первые годы внеутробной жизни.
Большинство диабетогенных вирусов вызывают аутоиммунный цитолиз островковых b-клеток. Действие аутоантител направлено против цитоплазматических и ядерных антигенов В-клеток. Эти аутоантитела способны связывать те же структуры клеток, что и панкреатотропные вирусы. Лимфотропные вирусы действуют как поликлональные инициаторы аутоиммунных механизмов (вирусы Эпштейна-Барр и кори) или в качестве инактиваторов Т-супрессоров (ретровирусы) либо стимуляторов Т-эффекторов. В этом случае аутоаллергический процесс может быть следствием вирус-индуцированного дефицита супрессоров и/или избытка эффекторов. В то же время иммунологический цитолиз присущ течению инфекций у наследственно предрасположенных субъектов.
Провоцирующая роль вирусов в генезе аутоиммунного цитолиза осуществляется через интерлейкины и интерфероны, особенно g-интерферон, при вирусном поражении поджелудочной железы. Эти цитокины вызывают экспрессию антигенов ГКГС на b-клетках и аутопрезентацию поверхностных антигенов b-клеток к последующему аутоиммунному цитолизу, а также появлению неоантигенов при персистирующих вирусных поражениях.
К химическим диабетогенам относятся аллоксан, мочевая кислота, стрептозоцин, дитизон, вакор (средства для борьбы с грызунами), бычий сывороточный альбумин (входит в состав коровьего молока), нитрозамины и нитрозомочевина (содержатся в копчёных продуктах), пентамидин (средство для лечения пневмоцистоза), продукты, содержащие пищевые цианиды (абрикосовое зерно, миндаль, африканский корнеплод кассава, которым питается около 400 млн. аборигенов, и др.). Курение и алкоголь способствуют повышению уровня цианидов в крови, усиливают проявления аутоиммунитета, способствуют развитию гемохроматоза и панкреатита.
В противовес диабетогенам описаны вещества с протекторным эффектом, так называемые антидиабетогены. Среди них называют серосодержащие аминокислоты, дефицит которых повышает токсичность пищевых цианидов, антиоксиданты, цинк (участвует в депонировании инсулина), витамин РР (тормозит процессы апоптоза и некроза, используется для лечения ИЗСД), полиненасыщенные жирные кислоты из морепродуктов (подавляют синтез широко известных ИЛ-1 и ФНО-α).
Основные механизмы химического повреждения панкреатических островков – это интерлейкин-зависимая экспрессия отсутствующих в норме на мембране b-клеток DR-белков, аутоиммунная альтерация и аутоаллергия, вызванная перекрёстными или общими антигенными детерминантами, а также иммунный ответ на экспрессию неоантигенов, обусловленный деструкцией b-клеток. В то же время возможно подавление пролиферации b-клеток антиклеточными антителами и медиаторами аутоиммунного воспаления.
Подводя итог сказанному относительно иммунных процессов ИЗСД, выделим главные. Это, во-первых, аллергический инсулит, вызванный цитотоксическими Т-лимфоцитами (клеточно-опосредованный тип аллергии) вследствие экспрессии на мембране b-клеток отсутствующих в норме DR-белков. Не исключена экспрессия неоантигенов – продуктов латентного вирусного генома, а также аномальная экспрессия генов ГКГС второго класса на b-клетках. Во-вторых, гуморально-опосредованный тип деструкции b-клеток, который представлен комплемент-зависимой и антитело-опосредованной клеточной цитотоксичностью (цитотоксический, или цитолитический, тип аллергических реакций). Выделяемые цитокины (ИЛ-1, ФНО-a, лимфотоксин, g-интерферон, фактор активации тромбоцитов, простагландины) ещё до выраженной аутоиммунной деструкции b-клеток приводят к торможению секреции инсулина. Особенно это относится к ИЛ-1, который снижает чувствительность b-клеток к глюкозе. Указанные цитокины, выделяемые лимфоцитами и макрофагами, обладают цитотоксическим, антипролиферативным и антисекреторным эффектами. Помимо аутоаллергического цитолиза, для ИЗСД характерно выключение митотической активности b-клеток.
Патогенез ИЗСД. Ключевое звено патогенеза ИЗСД – прогрессирующая гибель b-клеток панкреатических островков. Это приводит к изменению гетероклеточных взаимоотношений в островках, инсулинопении, избытку островковых и внеостровковых контринсулярных гормонов. В результате нарушаются утилизация глюкозы и все виды метаболизма. Хронические нарушения метаболизма порождают осложнения ИЗСД, главные из которых связаны с ангиопатиями.
Роль провоцирующего вирусного и/или химического диабетогена состоит в индукции аутоиммунной альтерации. У 10% больных с подтипом ИЗСД 1b (в сочетании с системной аутоиммунной полиэндокринопатией) провокация не является необходимой. У больных с подтипом ИЗСД 1а провоцирующее событие должно произойти в раннем онтогенезе или даже до рождения, т.к. ИЗСД – заболевание с длительным иммунологическим продромом и периодом метаболической компенсации. Интервал от дебюта аутоиммунного процесса до начала интолерантности к глюкозе составляет 3-4 года, а наиболее длительный период между первыми проявлениями снижения способности к выработке инсулина и явной метаболической декомпенсации – 1-12 лет. Пик заболеваемости ИЗСД приходится на возрастные периоды от рождения до 3 и от 9 до 13 лет. Морфофункциональная основа ИЗСД. В ответ на иммунологическую альтерацию в панкреатических островках развивается инсулит, проявляющийся гибелью b-клеток, экссудативными изменениями, инфильтрацией островков лимфоцитами, макрофагами, эозинофилами, извращением нейроваскулярных взаимоотношений, нарушениями топографии клеток и межклеточных контактов. К моменту формирования клинически явного диабета вес поджелудочной железы уменьшается в два, масса островков – в три раза, а β-клеток – более чем в 850 раз. В то же время в дезорганизованных островках растет доля А-клеток (до 75 %) и δ-клеток (до 25 %). В результате соотношение глюкагон/инсулин в крови больных ИЗСД по мере развития болезни стремится к бесконечности.
Еще раз подчеркнем, что ключевым звеном патогенеза ИЗСД является прогрессирующая гибель b-клеток вследствие аутоиммунной альтерации.
Однако генетическая предрасположенность лишь создает высокую вероятность заболевания. Для реализации необходимы инфекционные и неинфекционные диабетогенные факторы. Механизм действия диабетогенов связан с интерлейкин-зависимой экспрессией аутоантигенов b-клеток. Имеются основания предполагать, что значительная часть больных ИНСД – это лица, находящиеся в ранней стадии эволюции СД, но еще имеющие достаточно инсулина, чтобы предотвратить кетоацидоз. ИНСД у тучных имеет существенный патогенетический механизм – продукция адипоцитами контринсулярного цитокина ФНО-a.
Нарушения обмена веществ при СД
Проявлениями нарушения жирового обмена являются:
· гиперлипемия (содержание липидов в плазме выше 8 г/л, норма 4-8);
· гиперкетонемия (содержание кетоновых тел в плазме выше 30 мг/л или 520 мкмоль/л);
· гиперхолестеринемия (более 6 ммоль/л, норма 4,2-5,2);
· гиперфосфолипидемия (более 3,5 ммоль/л, норма 2,0-3,5);
· повышение содержания НЭЖК (более 0,8 ммоль/л);
· увеличение содержания триглицеридов – триглицеридемия (более 1,6 ммоль/л);
· увеличение содержания липопротеидов (более 8,6 г/л, норма 1,3-4,3).
Перечисленные показатели измененного жирового обмена обусловлены не только дефицитом инсулина, но и избытком контринсулярных гормонов, а также отсутствием липокаина. Гиперлипемия в отсутствии липокаина может приводить к жировой инфильтрации печени, чему способствуют:
· обеднение печени гликогеном;
· дефицит липотропных факторов, включая липокаин;
· жировая диета;
· избыток СТГ;
· инфекции и интоксикации.
Эти же факторы приводят к кетозу, однако, непосредственными причинами кетоза являются следующие:
· усиленный распад неэстерифицированных жирных кислот в печени;
· нарушение ресинтеза ацетоуксусной кислоты в высшие жирные кислоты;
· недостаточное окисление ацетоуксусной кислоты в цикле Кребса;
· повышенное образование ацетоуксусной кислоты в печени.
Вышеописанные изменения жирового обмена ведут к ускорению развития атеросклероза.
Нарушение белкового обмена. Эти нарушения касаются усиленного распада протеинов и ослабления синтеза белков. Торможение синтеза белка является предпосылкой образования из их компонентов углеводов – глюконеогенез, который стимулируется глюкокортикоидами и глюкагоном. Нарушается белковый состав плазмы:
· снижается содержание альбуминов,
· растет концентрация глобулинов,
· повышается уровень альфа-2-гликопротеидов.
Сахарный диабет второго типа, понятие, этиология, патогенез, последствия
ИНСД – это гетерогенная по этиологии и патогенезу группа заболеваний, характеризующаяся мультифакториальной наследственной предрасположенностью, относительной инсулиновой недостаточностью и инсулинорезистентностью. До сих пор дебатируется вопрос о том, что первично в патогенетической основе ИНСД – длительное действие выявляющих его факторов или возрастная экспрессия предрасполагающих генов. Поскольку доказано, что у большей части больных инсулинонезависимый сахарный диабет сочетается с ожирением и коррелирует с пожилым возрастом, значительная их часть имеет характерную комбинацию расстройств, объединённых в единый синдром – метаболический Х-синдром. К нему относят ГЛП IV или V типов, ускоренное развитие атеросклероза, ожирение андроидного типа, стеатоз печени, гипертензию, гиперурикемию, нефропатию.
Семейный риск ИНСД значителен – до 40 % сибсов и треть потомства у больных имеет толерантность к глюкозе. В отличие от ИЗСД, для ИНСД выявлены регионы, где проявляются эффект родоначальника и последствия инбридинга (остров Науру, где СД больны 83 % коренных жителей и район расселения индейцев пима, в Северной Америке, среди которых до 86 % больных СД). Несмотря на это, лишь для отдельных моногенных менделирующих разновидностей ИНСД установлен точный тип наследования.
Основные патогенетические звенья ИНСД представлены следующими дефектами:
· недостаточная секреция инсулина для утилизации глюкозы, особенно в течение первого часа после поступления углеводов, из-за снижения чувствительности глюкорецепторов В-клеток островков к глюкозо-стимулу;
· инсулинорезистентность вследствие аномалий молекулы инсулина, связывания инсулина циркулирующими в крови антителами к нему, наличием антител к инсулиновым рецепторам в клетках, уменьшением их числа и т.п.;
· усиленным образованием глюкозы печенью на протяжении полных суток, тогда как в норме синтез глюкозы происходит только днем.
· недостаточный ответ b-клеток на глюкозный сигнал без их деструкции,
· синтез «неправильного» проинсулина, либо нарушение его протеолиза,
· избыток циркулирующих антагонистов инсулина,
· аномалии инсулиновых рецепторов,
· блокада инсулиновых рецепторов,
· пострецепторный блок на разных уровнях в клетках-мишенях.
Все эти дефекты, вероятно, генетически детерминированы, причем большинство из них – моногенно. В отдельности подобные механизмы встречаются редко, в сумме эта гетерогенная группа составляет большую часть больных с диагнозом ИНСД. В доказательство высказанного приводим примеры случаев ИНСД при генетически обусловленных отдельных дефектах, составляющих комплекс патогенетических механизмов и ИНСД. Так, при диабете взрослых в юности (один из подтипов ИНСД) выявляется наследственный дефект фермента глюкокиназы (ген в коротком плече хромосомы 7). Этот фермент обусловливает чувствительность b-клеток и гепатоцитов к уровню глюкозы. Как следствие, данный дефект вызывает относительную «глухоту» b-клеток к глюкозному стимулу. Другим характерным проявлением ИНСД является скорее нарушение синтеза гликогена (гликогенез), чем захват глюкозы клетками и её окисление. Этот дефект наблюдается даже у нетучных родственников многих больных ИНСД. Это указывает на взаимосвязь с аллелью гликогенсинтетазы А2 и высокой вероятностью развития ИНСД (30 % частоты против 8 % в контрольной группе).
Аномалии самой молекулы инсулина могут возникать в результате мутации его структурного гена, кодирующего синтез биологически дефектных молекул этого гормона. Описаны единичные случаи ИНСД, обусловленного неполным превращением проинсулина в инсулин в ходе протеолиза, происходящего в грануле b-клетки. В норме только около 5 % секреторного продукта b-клеток представлено проинсулином, а биологическая активность последнего много меньше, чем инсулина. Больные такой формой ИНСД имеют дефект структурного гена проинсулина в точке отщепления терминального С-пептида. Это препятствует превращению проинсулина в инсулин и терминальный С-пептид. В крови таких больных радиоиммунологическим методом выявляется гиперинсулинемическое состояние.
Признаётся участие в патогенезе ИНСД генов-транспортёров глюкозы, в частности GluT-2, контролирующих поступление глюкозы в гепатоциты и панкреатоциты, или GluT-4 – в липоциты и миоциты. У некоторых пациентов ИНСД имеется избыточная экспрессия гена Rad, участвующего в работе по системе внутриклеточной передачи гормональных сигналов. Его продукт ингибирует инсулинозависимую активность транспортера глюкозы GluT-4.
Велика роль генетических аномалий возможного контринсулярного фактора b-клеток – амилина, или известного под другим названием амилоидогенного пептида. В норме амилин представляет собой пептид из 37 аминокислот и секретируется в небольших количествах вместе с инсулином. У многих больных ИНСД избыток амилина откладывается в синусоидных пространствах вокруг b-клеток, формируя в последующем амилоидные отложения, а синтез амилина растет параллельно секреции инсулина, что снижает ответ b-клеток на глюкозу и в последующем может вызвать ограничение их секреторных возможностей.
У некоторых больных ИНСД обнаружены особенности гена (хромосома 19), контролирующие пептиды инсулинового рецептора в клетках-мишенях. Так, аномальный пептид инсулинового рецептора может служить маркером ИНСД до четверти случаев, а тучные больные имеют низкую тирозинкиназную активность в клетках-мишенях инсулина. У 15 % больных имеется генетическая вариация в пострецепторном блоке клеток-мишеней, например, в структуре внутриклеточного инсулинового посредника IRS-1. У других больных описан избыточный синтез a2-SH-гликопротеина, который блокирует тирозинкиназную активность инсулиновых рецепторов клеток-мишеней. Выделен мембранный гликопротеин, ингибирующий тирозинкиназную активность рецепторов инсулина. Таким образом, чаще всего множественные пострецепторные дефекты могут вызывать первичную резистентность b-клеток к глюкозе, а клеток-мишеней инсулина – к этому гормону.
Экзогенным фактором, выявляющим генетические аномалии при ИНСД, служит переедание. Переедание повышает нагрузку на систему инсулиновой регуляции метаболизма. Формирующееся при этом ожирение вызывает включение дополнительных механизмов инсулинорезистентности. Низкая эффективность инсулина, характерная для ИНСД, теоретически могла бы компенсироваться гиперфункцией b-клеток. Однако при данном заболевании имеетместо пониженная их чувсивительность к глюкозе, в основе которой лежат разнообразные причины.
Для ИНСД характерны следующие особенности морфологии и функции островковых клеток:
· увеличение общего объема островковой ткани за счет возрастания числа инсулин-продуцирующих b-клеток,
· нередко амилоидоз и фиброз островков,
· резко уменьшено количество соматостатин-продуцирующих δ-клеток, что не может оказывать адекватного тормозного паракринного влияния на глюкагон-продуцирующие А-клетки,
· базальная секреция инсулина при ИНСД, как минимум, нормальна.
Патогенез ИНСД. Как уже упоминалось, в патогенезе ИНСД признается представление о первичности инсулинорезистентности и нарушении отвечаемости b-клеток на глюкозо-стимул. Это ведет к трехфазной картине течения ИНСД:
· стадия начальной инсулинорезистентности и компенсации гликемии.
· стадия выраженной инсулинрезистентности и относительной инсулиновой недостаточности, интолерантности к глюкозе.
· стадия снижения инсулиновой секреции и явного диабета.
Формирующаяся у больных прогрессирующая тучность усиливает инсулинрезистентность. Для ИНСД кетоацидоз не характерен по тем же причинам, по которым имеется ожирение – вследствие наличия инсулина, который сдерживает липолиз и создает условия для утилизации ацетил-КоА по пути стероидогенеза и липогенеза. Содержание малонил-КоА остается высоким, угнетая окисление жирных кислот и перекрывая путь кетогенеза. Метаболической платой за это служат тенденции к ожирению и крайне высокой гиперхолестеринемии. У больных ИНСД гиперхолестеринемия более умеренная.
Патогенез усиления инсулинрезистентности по мере ожирения (особенно гипертрофический и андроидный типы ожирения) обусловлен:
· уменьшаением плотности рецепторов инсулина на поверхности жировых клеток, что затрудняет гормон-рецепторное взаимодействие (ограничение экспрессии инсулиновых рецепторов).
· Торможение выделения адипоцитами цитокина, обладающего способностью снижать киназную активность рецептора инсулина в миоцитах и липоцитах – ФНО-a
Биологический смысл этого состоит в ограничении притока в адипоциды субстрата для липогенеза, тем более что названный цитокин является аппетит-подавляющим регулятором, откуда его второе название – кахексин. Но у тучных лиц кахексиновый механизм подавления неэффективен. В то же время ФНО-a вызывает дополнительную инсулинрезистентность, а также цитотоксичен для b-клеток, что открывает патогенетические пути перехода ИНСД во вторую и третью фазы развития. Однако ожирение не является единственно значимым фактором инсулинрезистентности. Важной причиной инсулинрезистентности могут быть адаптивное снижение экспрессии инсулиновых рецепторов при «сытом» состоянии клеток, а также иммунологические реакции.
Перечисленные механизмы ответственны за прогрессирование болезни. В третьей стадии ИНСД нет нарастания инсулинорезистентности, но уменьшаются секреторные возможности b-клеток. Опыты с удалением панкреас показывают, что 10 % массы интактных b-клеток достаточно для поддержания нормогликемии без соблюдения диеты. Предполагается, что инсулинорезистентность приобретает при ИНСД решающее значение только при нарушении отвечаемости b-клеток на глюкозу вследствие их дефекта. По этой причине подавляющее большинство лиц, страдающих тучностью и периферической инсулинрезистентностью, имеют гиперинсулинемию, но не гипергликемию. При наличии инсулинрезистентности компенсаторно развивается состояние гиперинсулинизма. Однако для стимуляции дефектных b-клеток требуется высокое содержание глюкозы, превосходящие почечный порог. Из-за глюкозурии гипергликемия не может увеличиваться бесконечно. При гипергликемии в 250 мг/дл и выше глюкоза теряется, а секреторные резервы b-клеток не могут быть отмобилизированы полностью. Состояние патологического равновесия достигается при больших или меньших концентрациях инсулина в зависимости от преобладания нсулинорезистентности или дефекта b-клеток, но инсулина всегда недостаточно по отношению к имеющемуся избытку глюкозы в крови. Тяжелый кетоацидоз, который не купируется инсулином, связывают с нсулинорезистентностью. Даже «малые дозы» инсулина могут вызвать повышение его содержание в крови в 4-15 раз, не оказывая терапевтического эффекта. Это послужило основанием для заключения, что нсулинорезистентность в условиях кетоацидоза обусловлена избытком протонов и СЖК.
Основные осложнения при сахарном диабете, этиология, патогенез, проявления
К острым осложнениям относятся коматозные состояния, к хроническим – микроангиопатии, макроангиопатии (МиП и МаП), инсулинорезистентность, нейропатия, нефропатия, иммунодефициты. Для ИНСД более характерны гиперосмолярная и гиперлактацидемическая комы.
МаП встречаются чаще и проявляются хронической ишемической болезнью сердца, нарушением мозгового кровообращения и облитерирующим атеросклерозом артерий нижних конечностей. В патогенезе МаП ведущее значение имеет ускоренное развитие атеросклероза, тогда как при МиП – гипергликемия. Механизмы ускоренного развития атеросклероза множественны – гиперлипопротеинемия, гипертензия, гипергликемия, гиперинсулинизм, тромбофилитический синдром.
Патогенез острых осложнений сахарного диабета. Кетоацидотическая кома. По мере развития СД все пути использования избытка ацетил-КоА блокируются, за исключением тех, которые ведут к кетозу и синтезу холестерина, метаболическому ацидозу, потере воды и электролитов, гемоконцентрации, недостаточности периферического кровообращения, аритмиям, шоку. Развивается компенсаторный метаболический ацидоз с потерей натрия мочой и компенсаторным выходом из клеток протона, что усугубляет ацидоз. Вследствие глубокой гипоксии ЦНС функции пневмотаксического центра замещаются гаспинг-центром, развивается дыхание Куссмауля, гипервентиляция, гипокапния, гипобикарбонатемия, что углубляет ацидоз.
Вследствие гипоксии в ткани мозга накапливается избыток лактата, что ведет к усугублению ацидоза. Ацидоз по типу порочного круга при диабетической коме вызывает усиление инсулинорезистентности, так как инсулин в кислой среде теряет сродство к своему рецептору. Кроме того, инсулинорезистентность обусловлена высоким уровнем СЖК и высвобождением контринсулярных гормонов – антагонистов инсулина (адреналина, глюкокортикоидов, глюкагона, вазопрессина). Диабетическая (кетонемическая, ацидотическая) кома обусловлена токсическим влиянием кетоновых тел и тканевой гипоксии на клетки ЦНС, обезвоживанием, ацидозом. Усиленный катаболизм белка приводит к увеличению содержания аммиака и мочевины, продукционной гиперазотемии, что углубляет интоксикацию и гипоксию мозга. Гипоксия нейронов приводит к расстройству дыхания, сосудистому коллапсу, снижению мышечного тонуса, нарушению ВНД.
Лактоацидоз и гиперлактацидемическая кома. Встречаются довольно часто (токсические дистрофии, циррозы печени), при сердечной недостаточности и других болезнях и нередко в тяжёлой форме – при декомпенсации ИНСД, который лечили бигуанидами – блокаторами глюконеогенеза.
В крови повышается уровень лактата более 5 ммоль/л при норме до 1,5 ммоль/л, значение рН артериальной крови 7,25 ед. и менее. Лактоацидоз является результатом гипоксии и физического переутомления. Клинически характерны дыхание Куссмауля, гипотензия, гипотермия, обезвоживание, ацидоз, циркуляторный коллапс, отсутствие кетонурии.
Гипергликемическая (гиперосмолярная) кома встречается реже кетоацидотической в основном у больных старше 50 лет, чаще беспомощных. Провоцируется дегидратацией организма (рвота, понос, лечение диуретиками, ограничение приема жидкости). Кетоацидоз отсутствует, гипергликемия может нарастать растянуто во времени до высоких цифр (55 ммоль/л и более). В патогенезе имеют значение следующие факторы:
· Гипергликемия 55-200 ммоль/л (1000-3600 мг/дл).
· Гипернатриемия, гиперхлоремия (обусловленные гиперальдостеронизмом в ответ на дегидратационную гиповолемию),
· Гиперазотемия (за счет мочевины) из-за ограничения диуреза.
· Отсутствие дыхания Куссмауля, запаха ацетона.
Патогенез хронических осложнений.
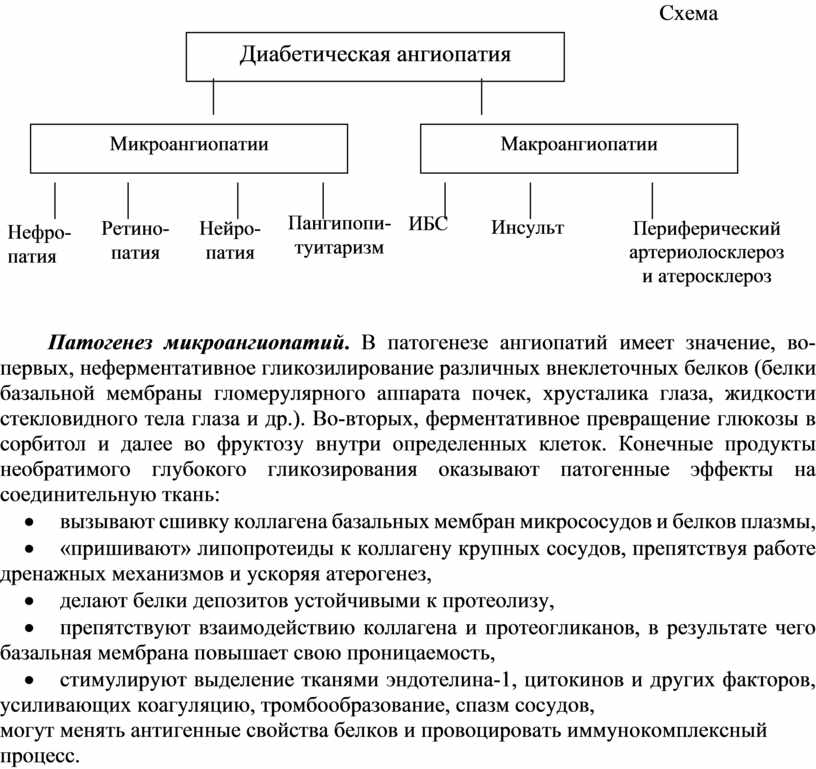
Диабетические ангиопатии являются главными осложнениями СД, инвалидизации и смерти больных (Схема). Понятие «ангиопатии» включает микроангиопатии (поражение капилляров, венул, артериол, прежде всего, их базальной мембраны) и макроангиопатии (поражение крупных артерий).
В течение СД любого типа наблюдаются комбинированная ангиопатия с преобладанием у молодых лиц ИЗСД типа микроангиопатии, у лиц старше 40 лет и ИНСД типа макроангиопатия с прогрессирующим развитием атеросклероза. Общим для МиП всех локализаций являются аневризмы капилляров, утолщение стенок артериол, капилляров, венул за счет накопления в базальной мембране гомогенных или слоистых веществ, пролиферация эндотелия в просвет сосудов (вплоть до полной облитерация), тучноклеточная реакция в периваскулярной ткани. Так, например, ИЗСД – главная причина слепоты и одна из ведущих системных причин ХПН.
Схема
 |
||||||||||||||||||
|
|
|||||||||||||||||
 |
 |
 |
 |
 |
 |
|||||||||||||
Патогенез микроангиопатий. В патогенезе ангиопатий имеет значение, во-первых, неферментативное гликозилирование различных внеклеточных белков (белки базальной мембраны гломерулярного аппарата почек, хрусталика глаза, жидкости стекловидного тела глаза и др.). Во-вторых, ферментативное превращение глюкозы в сорбитол и далее во фруктозу внутри определенных клеток. Конечные продукты необратимого глубокого гликозирования оказывают патогенные эффекты на соединительную ткань:
· вызывают сшивку коллагена базальных мембран микрососудов и белков плазмы,
· «пришивают» липопротеиды к коллагену крупных сосудов, препятствуя работе дренажных механизмов и ускоряя атерогенез,
· делают белки депозитов устойчивыми к протеолизу,
· препятствуют взаимодействию коллагена и протеогликанов, в результате чего базальная мембрана повышает свою проницаемость,
· стимулируют выделение тканями эндотелина-1, цитокинов и других факторов, усиливающих коагуляцию, тромбообразование, спазм сосудов,
· могут менять антигенные свойства белков и провоцировать иммунокомплексный процесс.
· Скачано с www.znanio.ru













Материалы на данной страницы взяты из открытых источников либо размещены пользователем в соответствии с договором-офертой сайта. Вы можете сообщить о нарушении.
